

Targeting de novo lipid synthesis induces lipotoxicity and impairs DNA damage repair in glioblastoma mouse models
- PMID: 36652537
- PMCID: PMC9942236
- DOI: 10.1126/scitranslmed.abq6288
Targeting de novo lipid synthesis induces lipotoxicity and impairs DNA damage repair in glioblastoma mouse models
Abstract
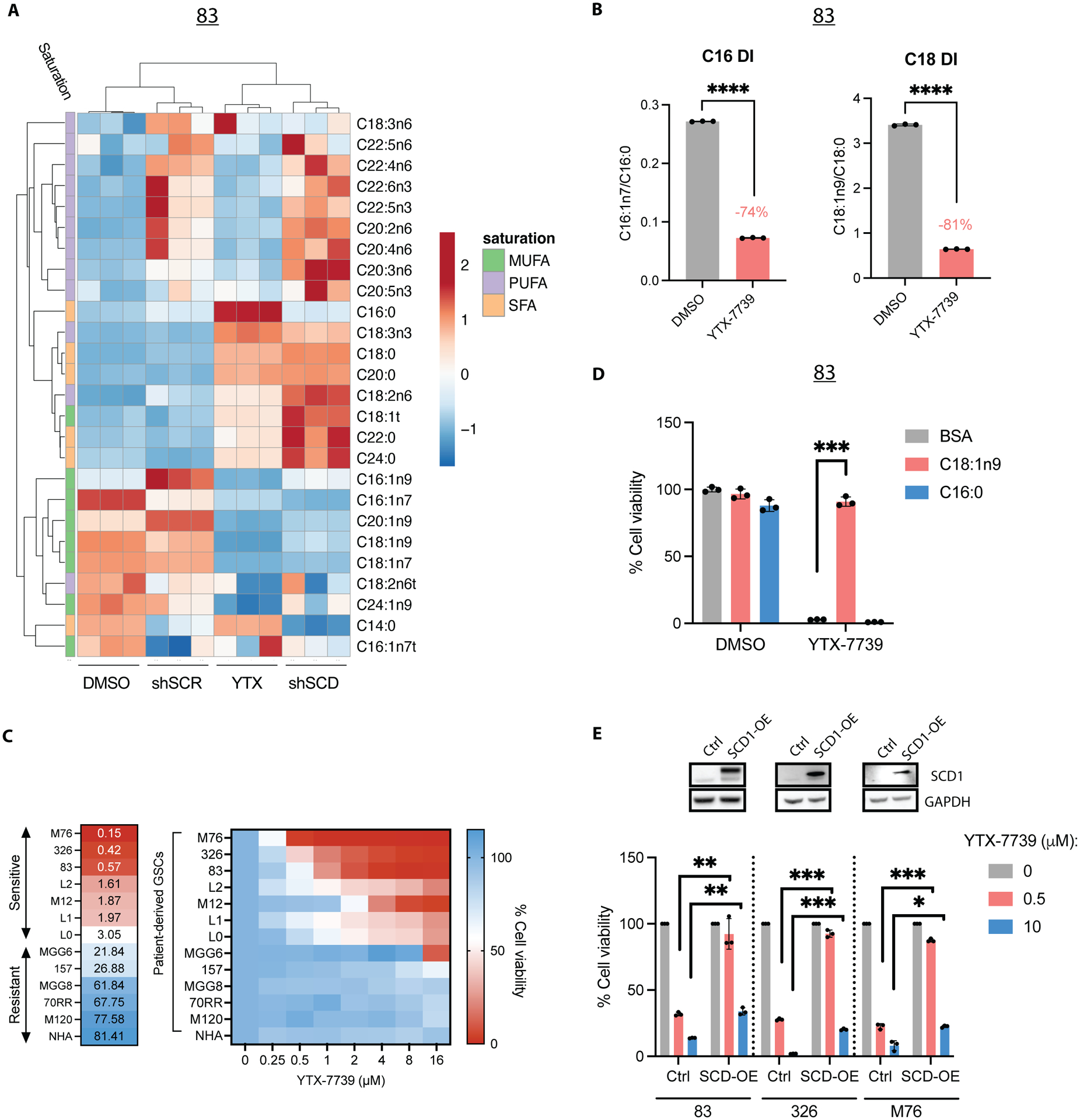
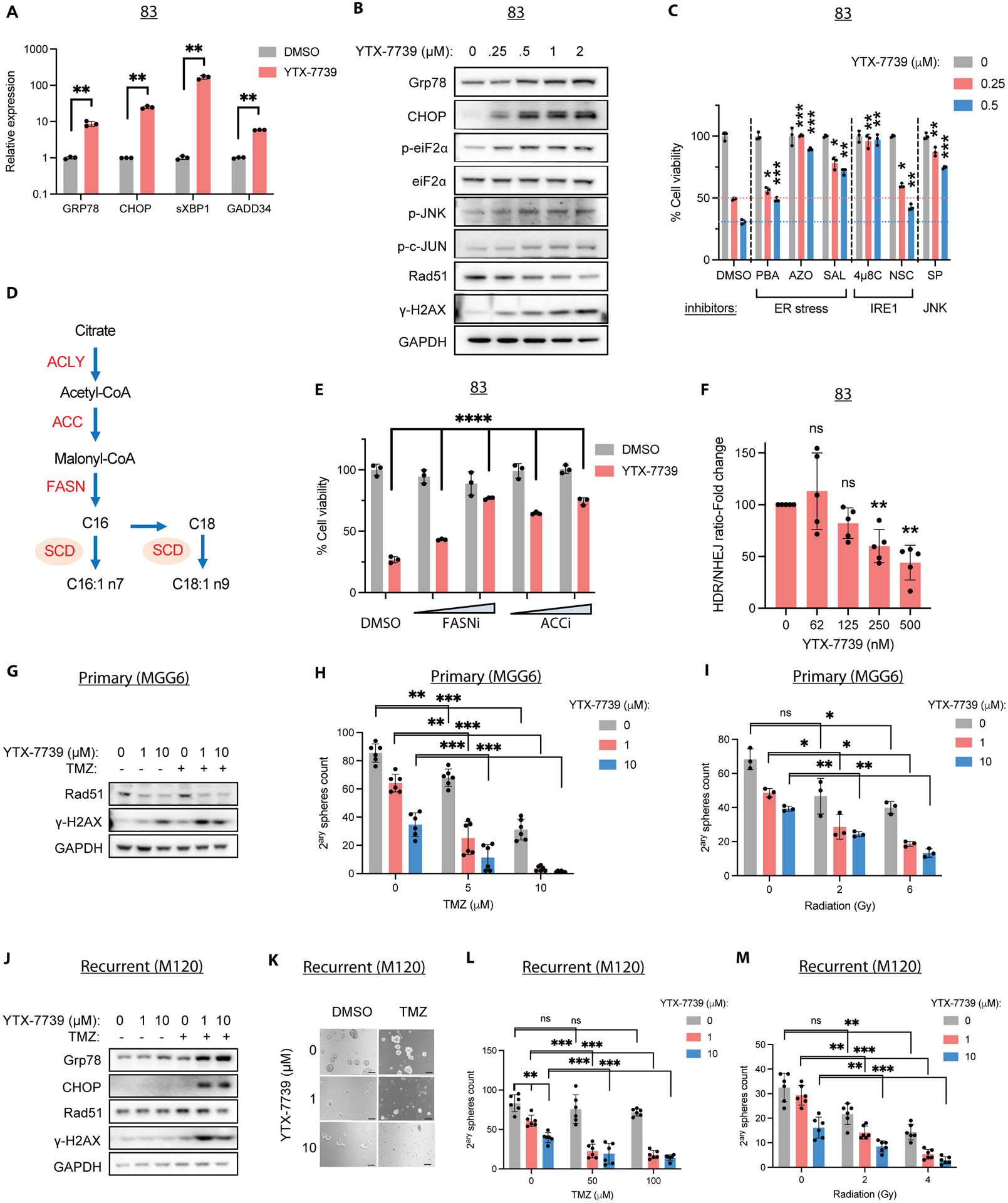
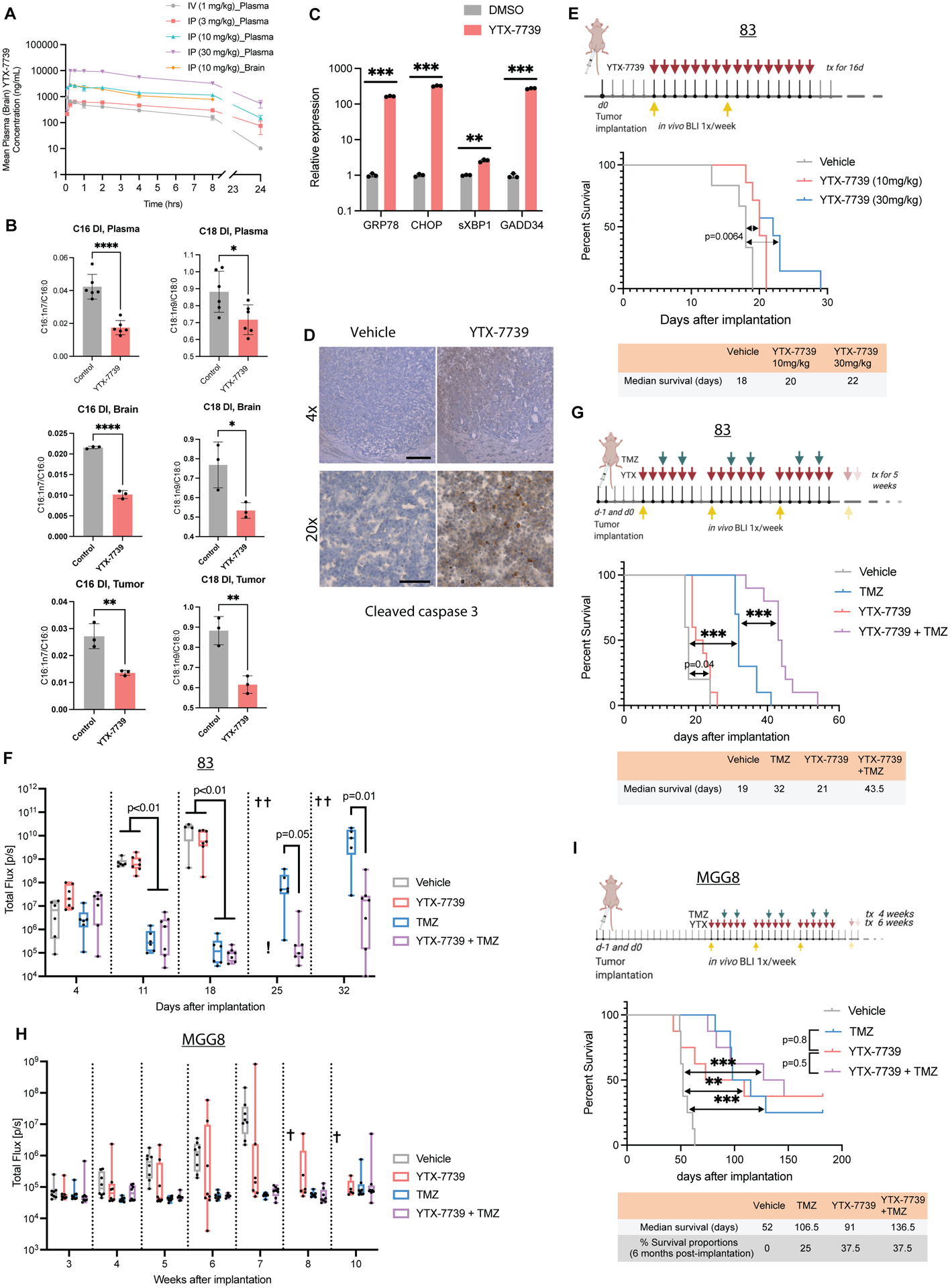
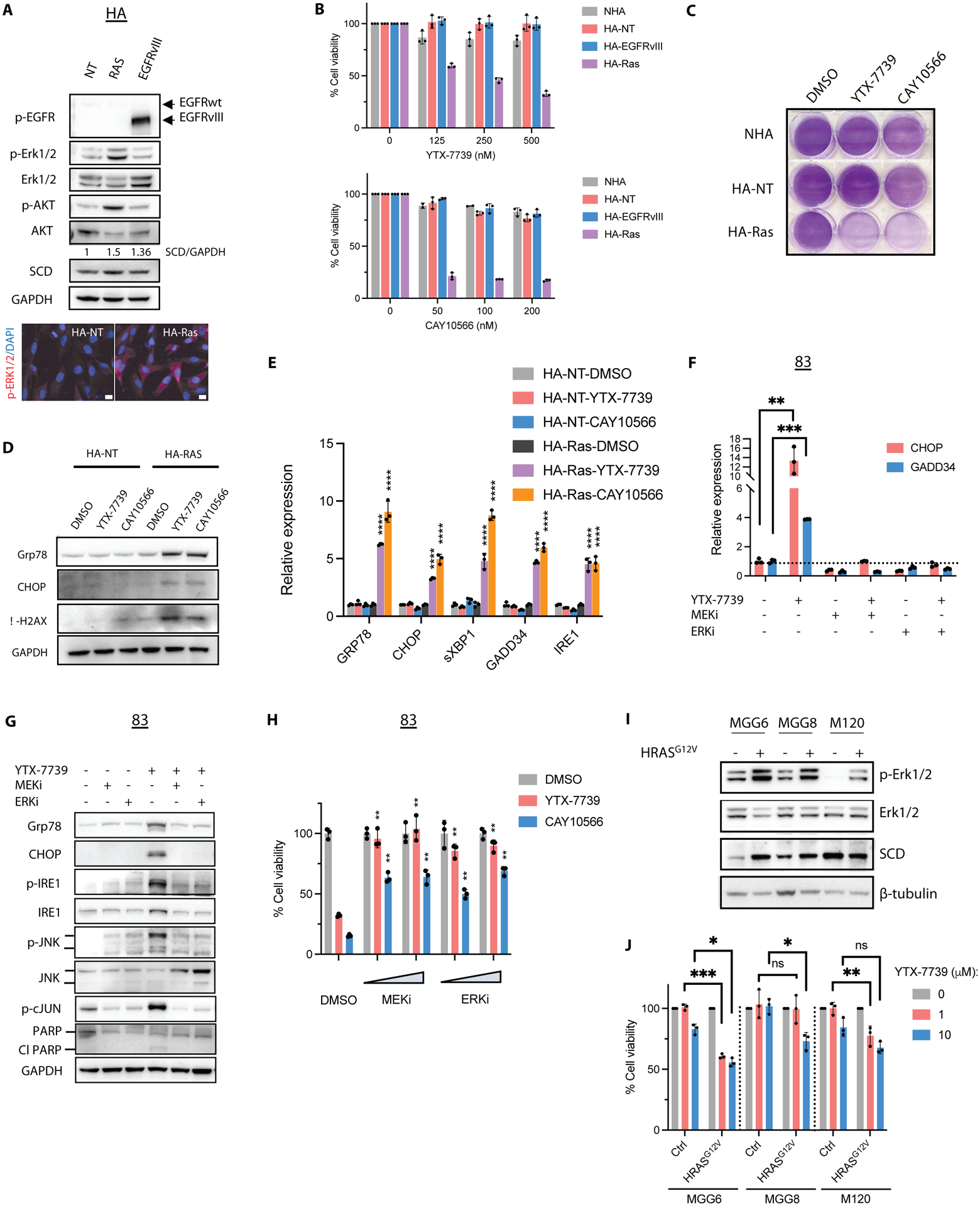
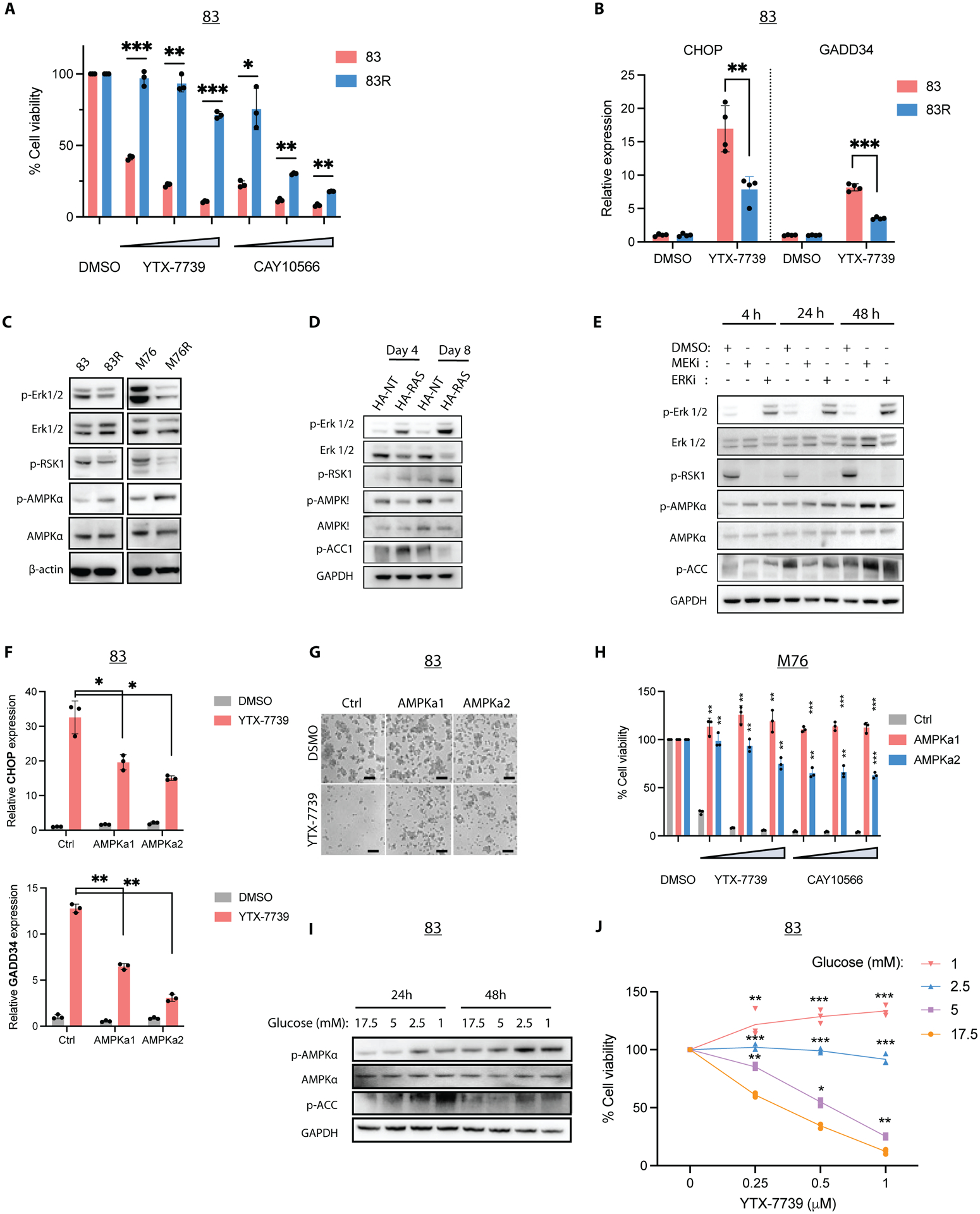
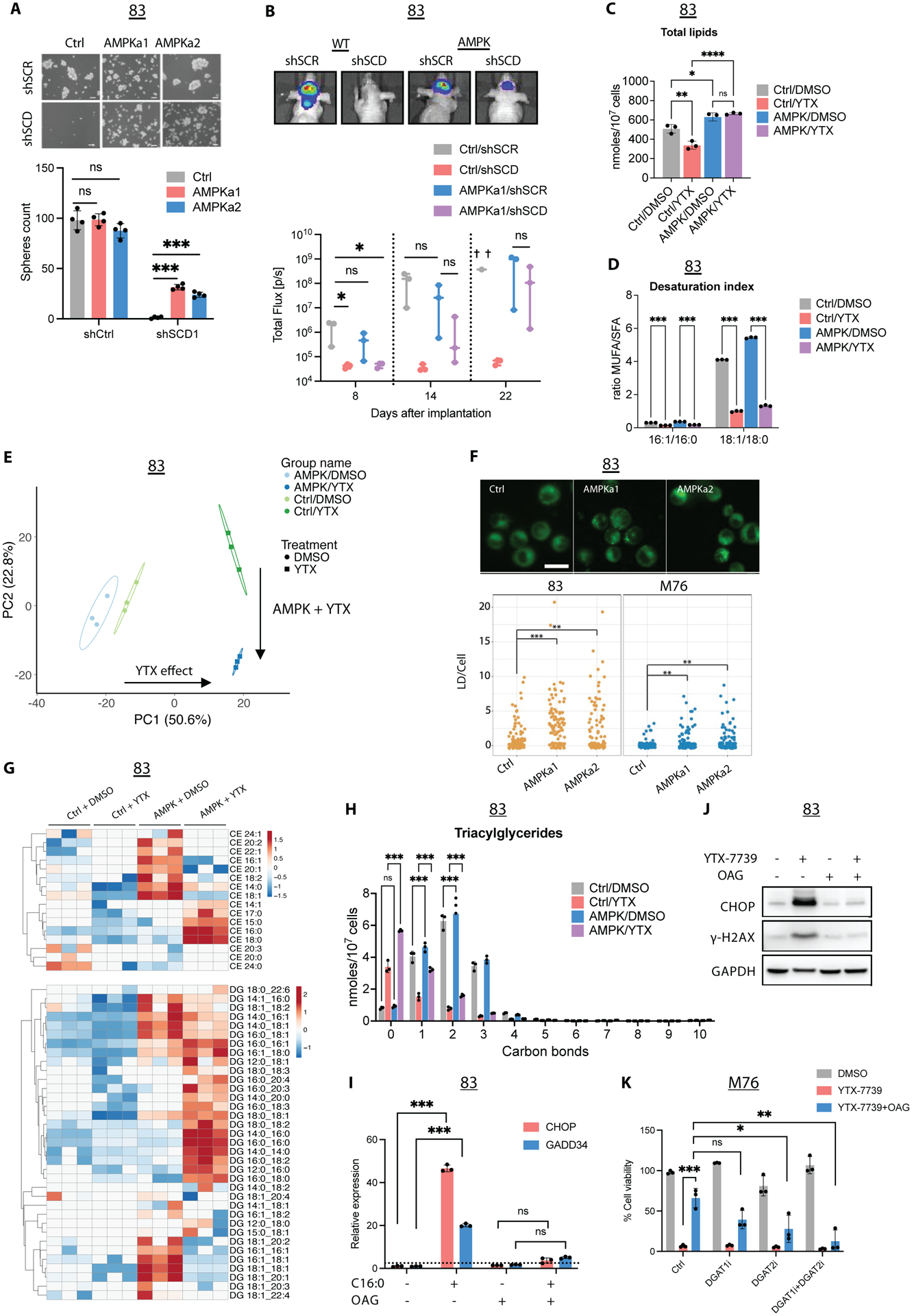
Deregulated de novo lipid synthesis (DNLS) is a potential druggable vulnerability in glioblastoma (GBM), a highly lethal and incurable cancer. Yet the molecular mechanisms that determine susceptibility to DNLS-targeted therapies remain unknown, and the lack of brain-penetrant inhibitors of DNLS has prevented their clinical evaluation as GBM therapeutics. Here, we report that YTX-7739, a clinical-stage inhibitor of stearoyl CoA desaturase (SCD), triggers lipotoxicity in patient-derived GBM stem-like cells (GSCs) and inhibits fatty acid desaturation in GSCs orthotopically implanted in mice. When administered as a single agent, or in combination with temozolomide (TMZ), YTX-7739 showed therapeutic efficacy in orthotopic GSC mouse models owing to its lipotoxicity and ability to impair DNA damage repair. Leveraging genetic, pharmacological, and physiological manipulation of key signaling nodes in gliomagenesis complemented with shotgun lipidomics, we show that aberrant MEK/ERK signaling and its repression of the energy sensor AMP-activated protein kinase (AMPK) primarily drive therapeutic vulnerability to SCD and other DNLS inhibitors. Conversely, AMPK activation mitigates lipotoxicity and renders GSCs resistant to the loss of DNLS, both in culture and in vivo, by decreasing the saturation state of phospholipids and diverting toxic lipids into lipid droplets. Together, our findings reveal mechanisms of metabolic plasticity in GSCs and provide a framework for the rational integration of DNLS-targeted GBM therapies.
Figures
Comment in
-
Drugging a metabolic vulnerability in glioblastoma.Nat Rev Drug Discov. 2023 Mar;22(3):182. doi: 10.1038/d41573-023-00020-0. Nat Rev Drug Discov. 2023. PMID: 36750686 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
