

Smarcd3 is an epigenetic modulator of the metabolic landscape in pancreatic ductal adenocarcinoma
- PMID: 36653361
- PMCID: PMC9849267
- DOI: 10.1038/s41467-023-35796-7
Smarcd3 is an epigenetic modulator of the metabolic landscape in pancreatic ductal adenocarcinoma
Abstract
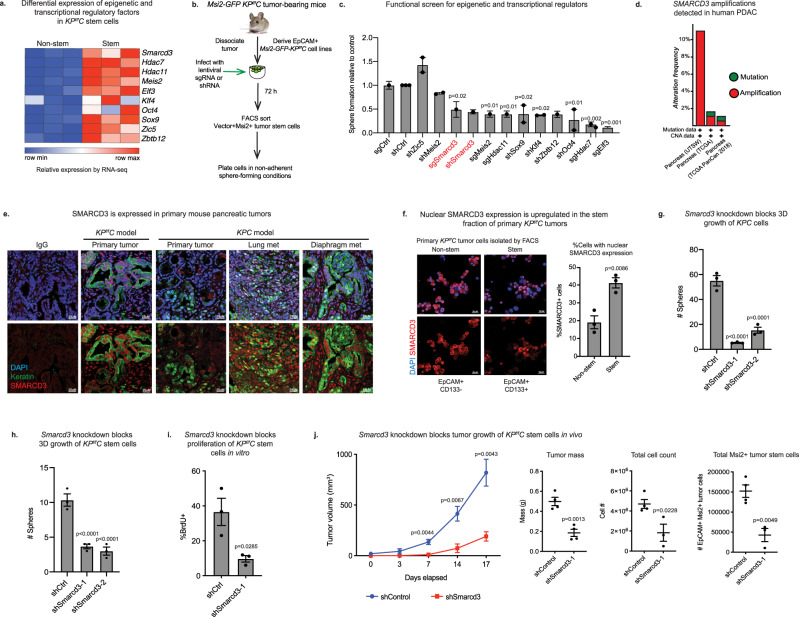
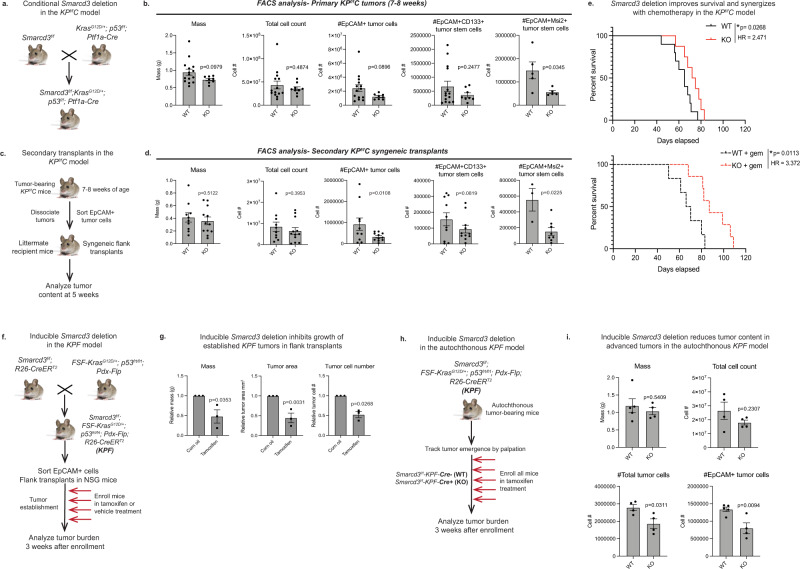
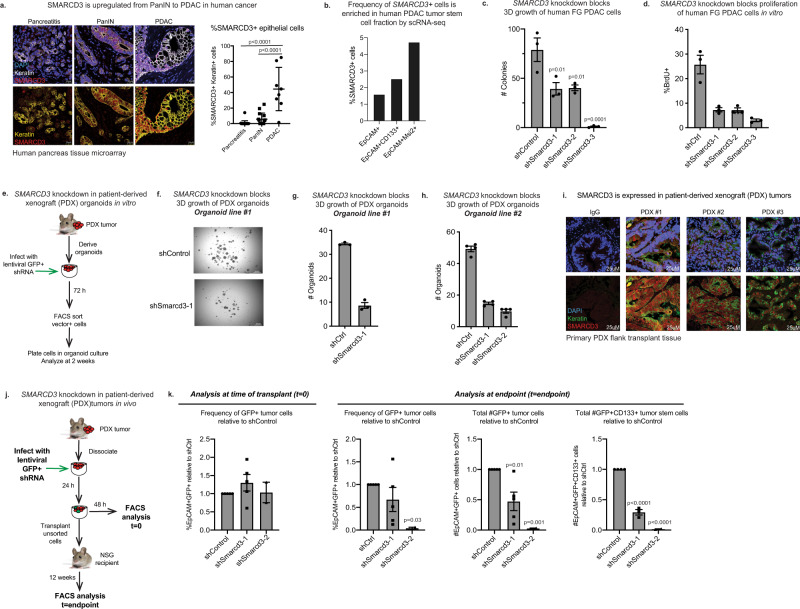
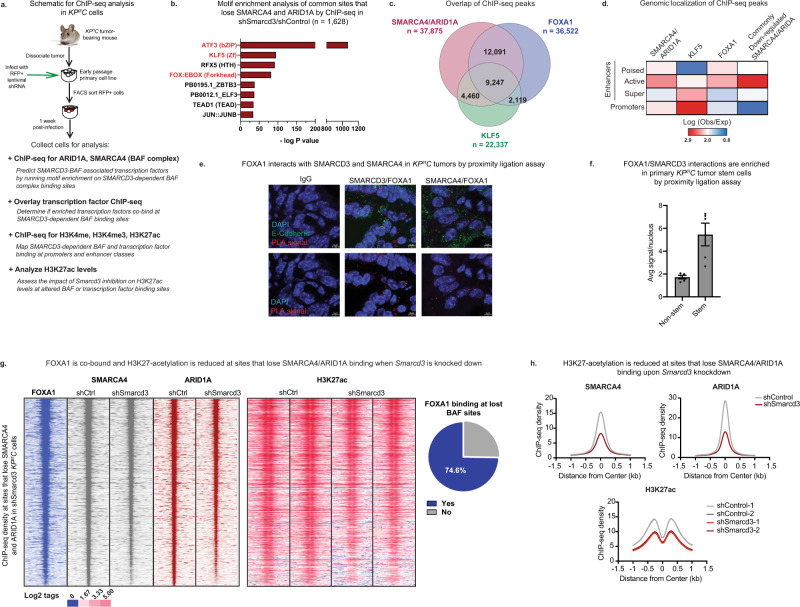
Pancreatic cancer is characterized by extensive resistance to conventional therapies, making clinical management a challenge. Here we map the epigenetic dependencies of cancer stem cells, cells that preferentially evade therapy and drive progression, and identify SWI/SNF complex member SMARCD3 as a regulator of pancreatic cancer cells. Although SWI/SNF subunits often act as tumor suppressors, we show that SMARCD3 is amplified in cancer, enriched in pancreatic cancer stem cells and upregulated in the human disease. Diverse genetic mouse models of pancreatic cancer and stage-specific Smarcd3 deletion reveal that Smarcd3 loss preferentially impacts established tumors, improving survival especially in context of chemotherapy. Mechanistically, SMARCD3 acts with FOXA1 to control lipid and fatty acid metabolism, programs associated with therapy resistance and poor prognosis in cancer. These data identify SMARCD3 as an epigenetic modulator responsible for establishing the metabolic landscape in aggressive pancreatic cancer cells and a potential target for new therapies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA. 2021;71:7–33. - PubMed
-
- Kleeff J, et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016;2:1–22. - PubMed
-
- Black, J. R. M. & McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer.21, 379–392 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
