

A streamlined tandem tip-based workflow for sensitive nanoscale phosphoproteomics
- PMID: 36653408
- PMCID: PMC9849344
- DOI: 10.1038/s42003-022-04400-x
A streamlined tandem tip-based workflow for sensitive nanoscale phosphoproteomics
Abstract
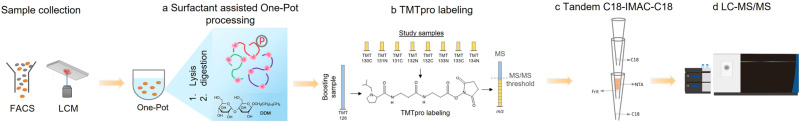
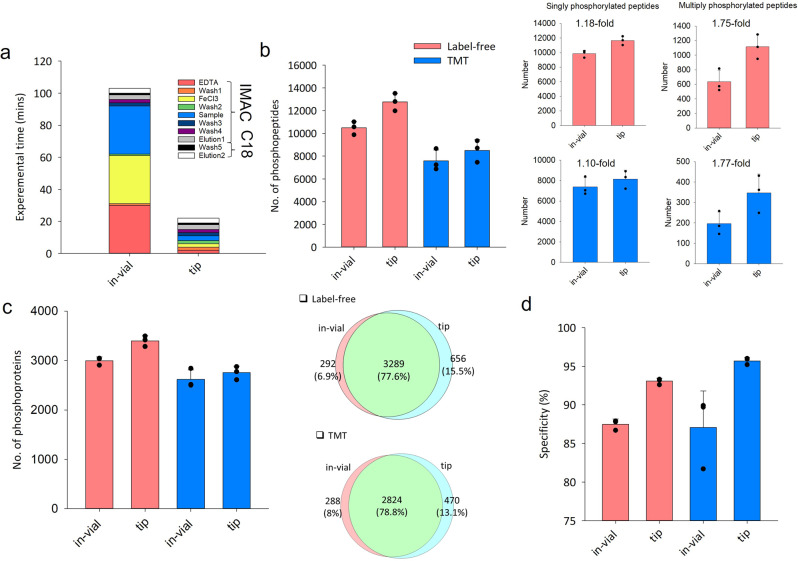
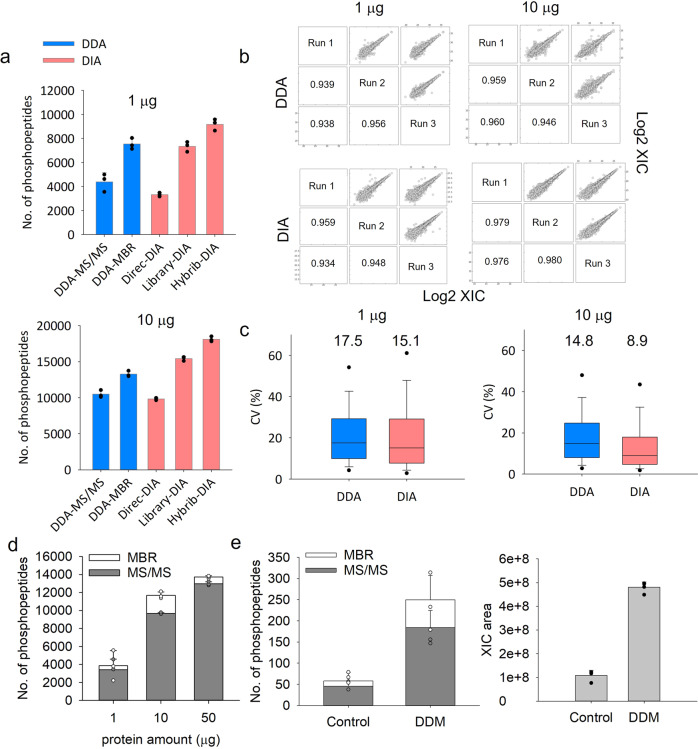
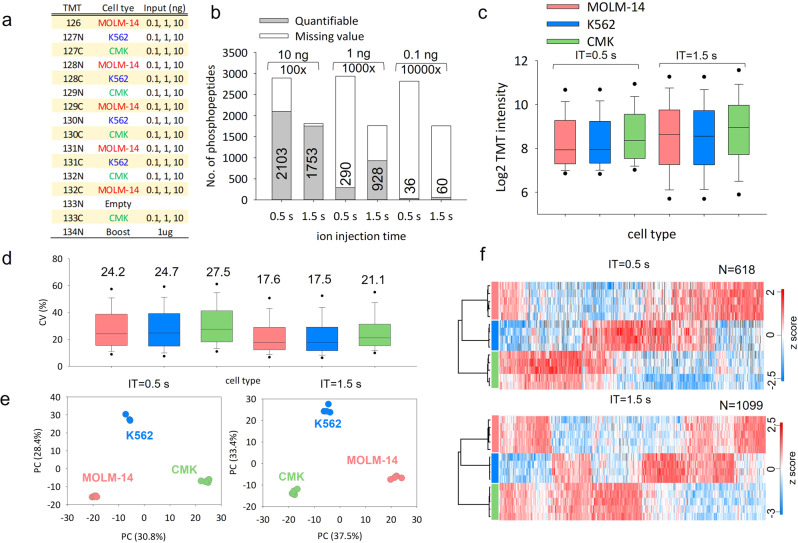
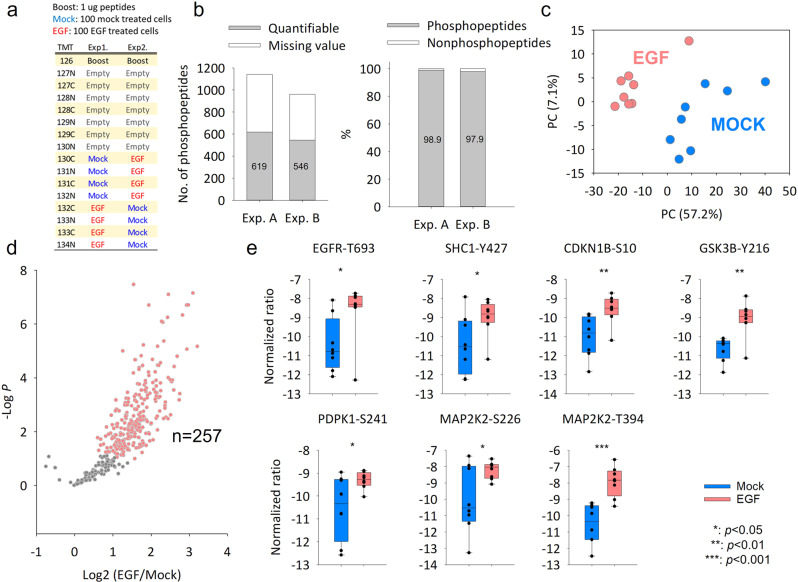
Effective phosphoproteome of nanoscale sample analysis remains a daunting task, primarily due to significant sample loss associated with non-specific surface adsorption during enrichment of low stoichiometric phosphopeptide. We develop a tandem tip phosphoproteomics sample preparation method that is capable of sample cleanup and enrichment without additional sample transfer, and its integration with our recently developed SOP (Surfactant-assisted One-Pot sample preparation) and iBASIL (improved Boosting to Amplify Signal with Isobaric Labeling) approaches provides a streamlined workflow enabling sensitive, high-throughput nanoscale phosphoproteome measurements. This approach significantly reduces both sample loss and processing time, allowing the identification of >3000 (>9500) phosphopeptides from 1 (10) µg of cell lysate using the label-free method without a spectral library. It also enables precise quantification of ~600 phosphopeptides from 100 sorted cells (single-cell level input for the enriched phosphopeptides) and ~700 phosphopeptides from human spleen tissue voxels with a spatial resolution of 200 µm (equivalent to ~100 cells) in a high-throughput manner. The new workflow opens avenues for phosphoproteome profiling of mass-limited samples at the low nanogram level.
© 2023. Battelle Memorial Institute, Hsu, Jorgenson, and Wasserfall.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
