

Mono- and biallelic variant effects on disease at biobank scale
- PMID: 36653560
- PMCID: PMC9849130
- DOI: 10.1038/s41586-022-05420-7
Mono- and biallelic variant effects on disease at biobank scale
Abstract
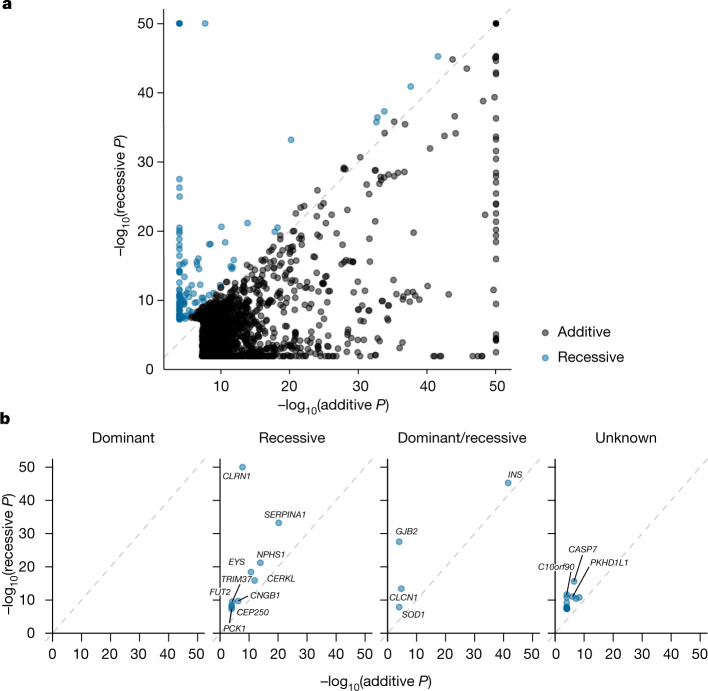
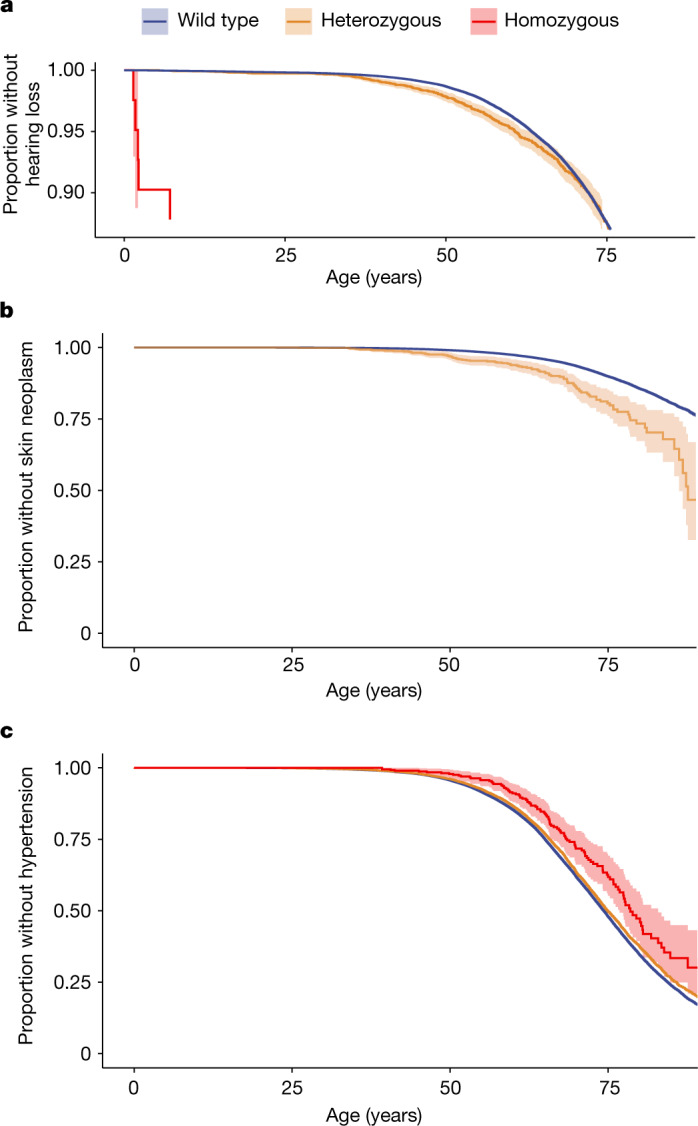
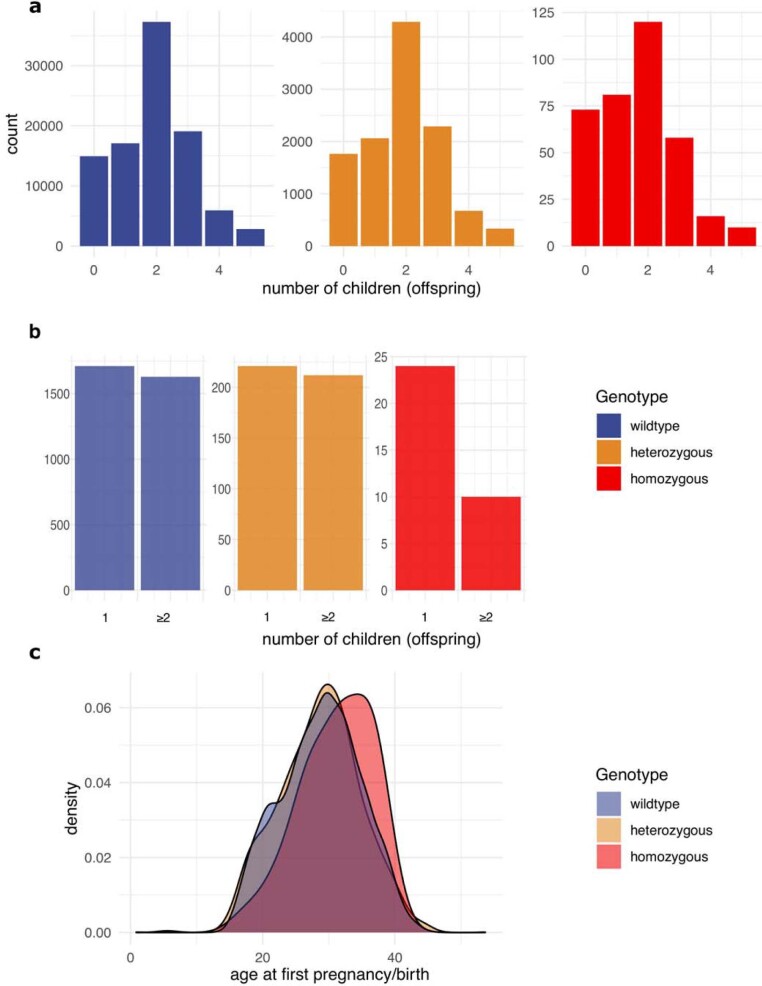
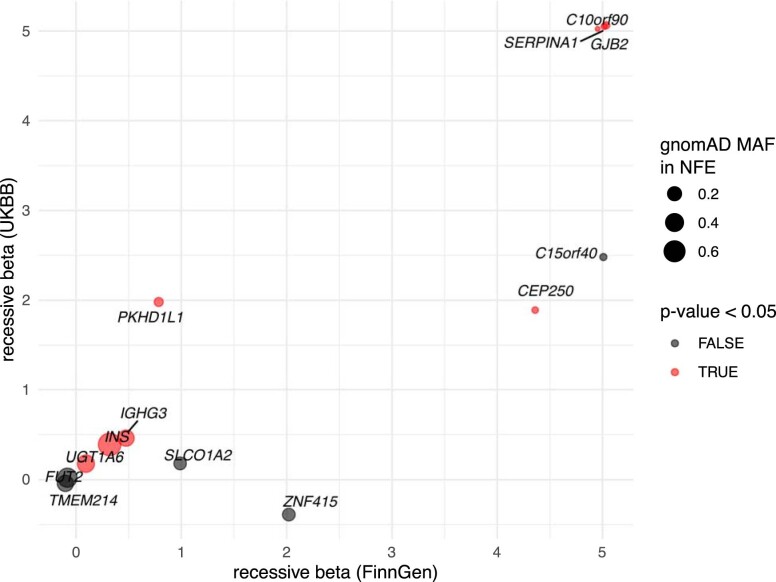
Identifying causal factors for Mendelian and common diseases is an ongoing challenge in medical genetics1. Population bottleneck events, such as those that occurred in the history of the Finnish population, enrich some homozygous variants to higher frequencies, which facilitates the identification of variants that cause diseases with recessive inheritance2,3. Here we examine the homozygous and heterozygous effects of 44,370 coding variants on 2,444 disease phenotypes using data from the nationwide electronic health records of 176,899 Finnish individuals. We find associations for homozygous genotypes across a broad spectrum of phenotypes, including known associations with retinal dystrophy and novel associations with adult-onset cataract and female infertility. Of the recessive disease associations that we identify, 13 out of 20 would have been missed by the additive model that is typically used in genome-wide association studies. We use these results to find many known Mendelian variants whose inheritance cannot be adequately described by a conventional definition of dominant or recessive. In particular, we find variants that are known to cause diseases with recessive inheritance with significant heterozygous phenotypic effects. Similarly, we find presumed benign variants with disease effects. Our results show how biobanks, particularly in founder populations, can broaden our understanding of complex dosage effects of Mendelian variants on disease.
© 2023. The Author(s).
Conflict of interest statement
A.P. is a member of the Pfizer Genetics Scientific Advisory Panel. M.J.D. is a founder of Maze Therapeutics. The remaining authors declare no competing interests.
Figures






Comment in
-
The FinnGen study: disease insights from a 'bottlenecked' population.Nat Rev Genet. 2023 Apr;24(4):207. doi: 10.1038/s41576-023-00584-y. Nat Rev Genet. 2023. PMID: 36747004 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
