

Identification and Characterization of ML321: A Novel and Highly Selective D2 Dopamine Receptor Antagonist with Efficacy in Animal Models That Predict Atypical Antipsychotic Activity
- PMID: 36654757
- PMCID: PMC9841785
- DOI: 10.1021/acsptsci.2c00202
Identification and Characterization of ML321: A Novel and Highly Selective D2 Dopamine Receptor Antagonist with Efficacy in Animal Models That Predict Atypical Antipsychotic Activity
Abstract
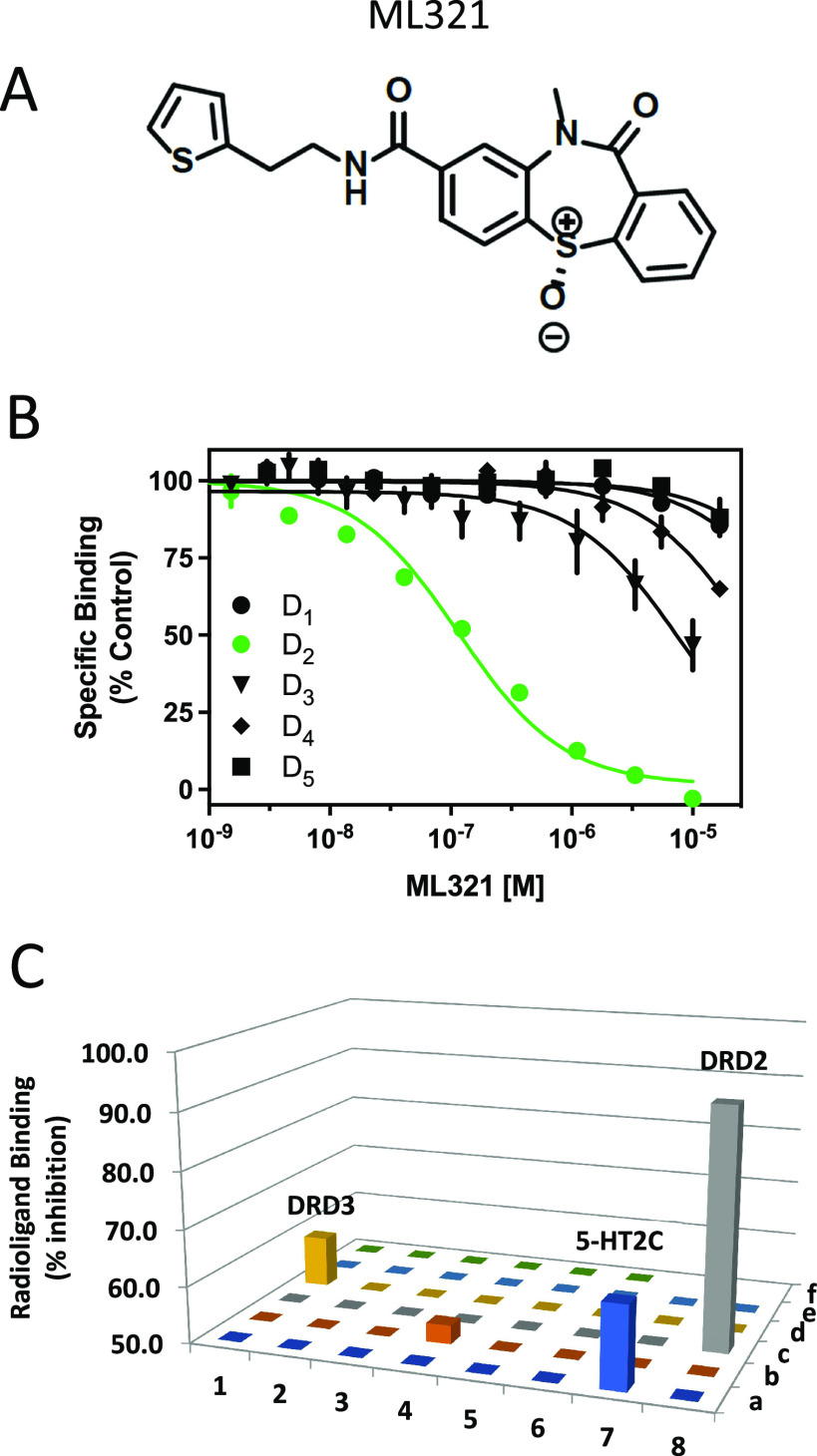
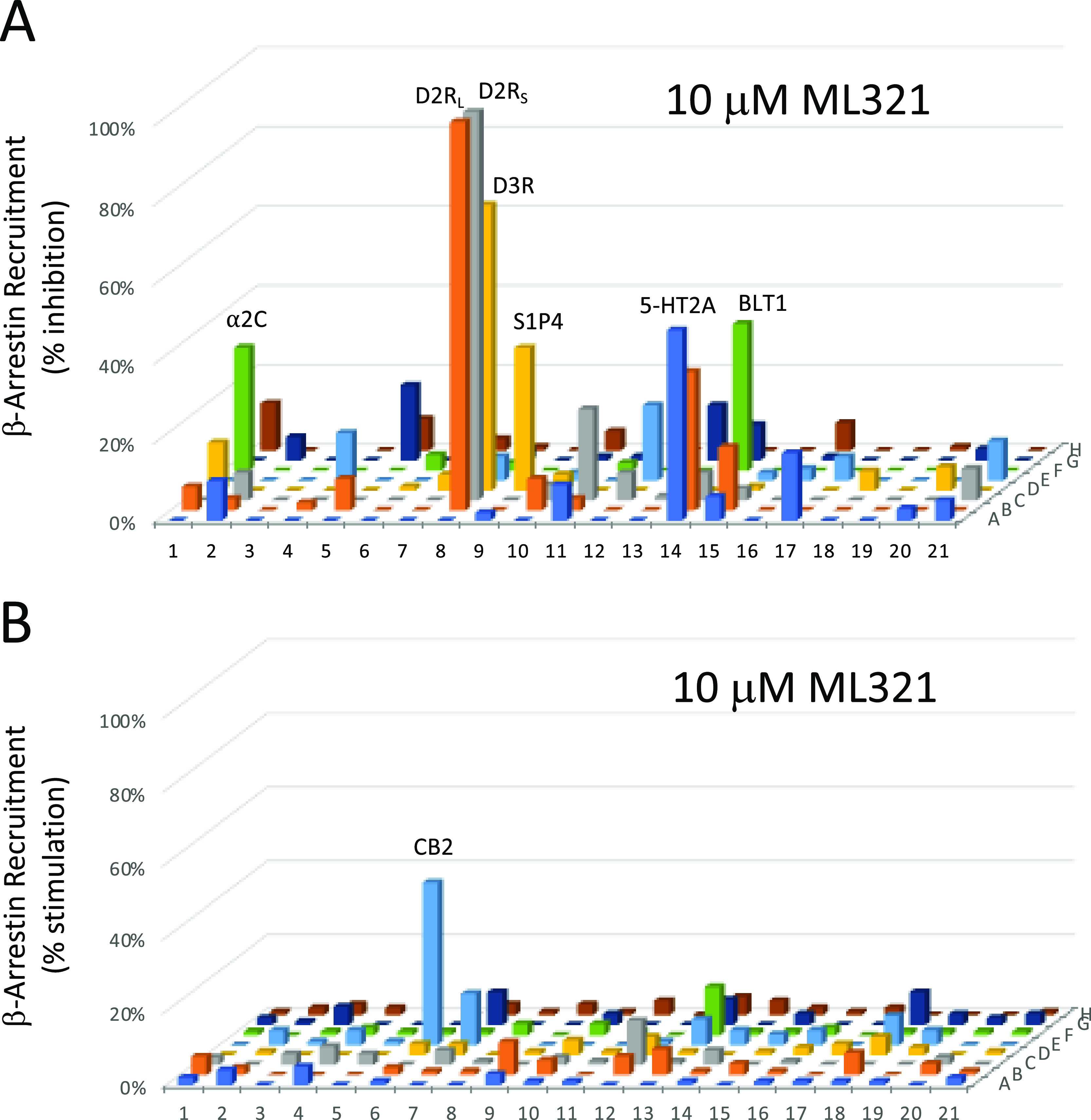
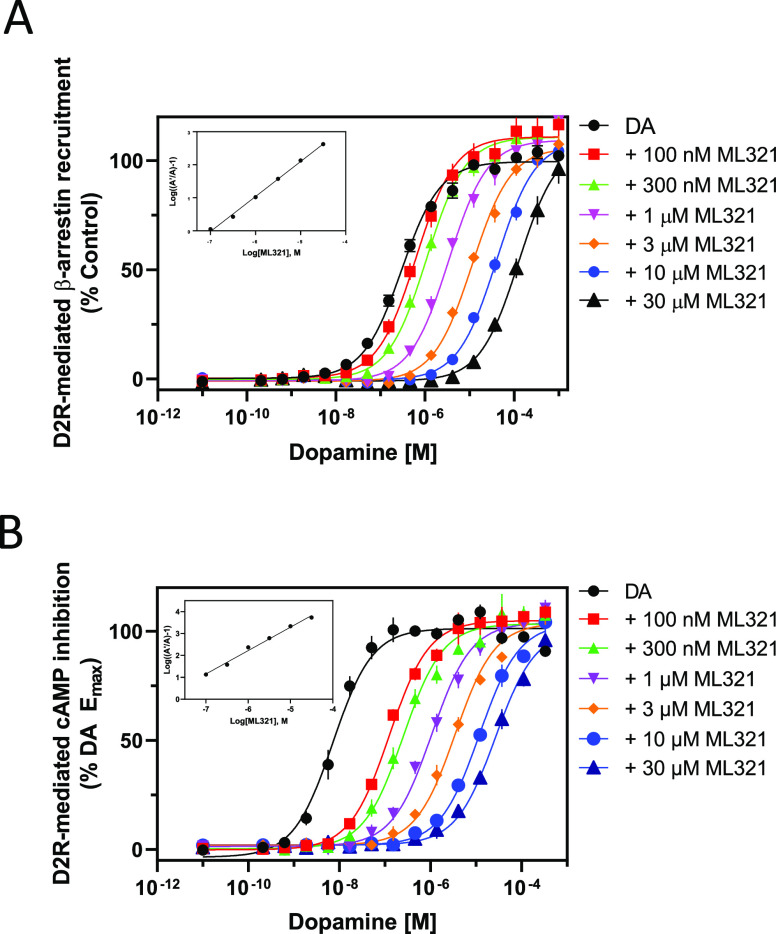
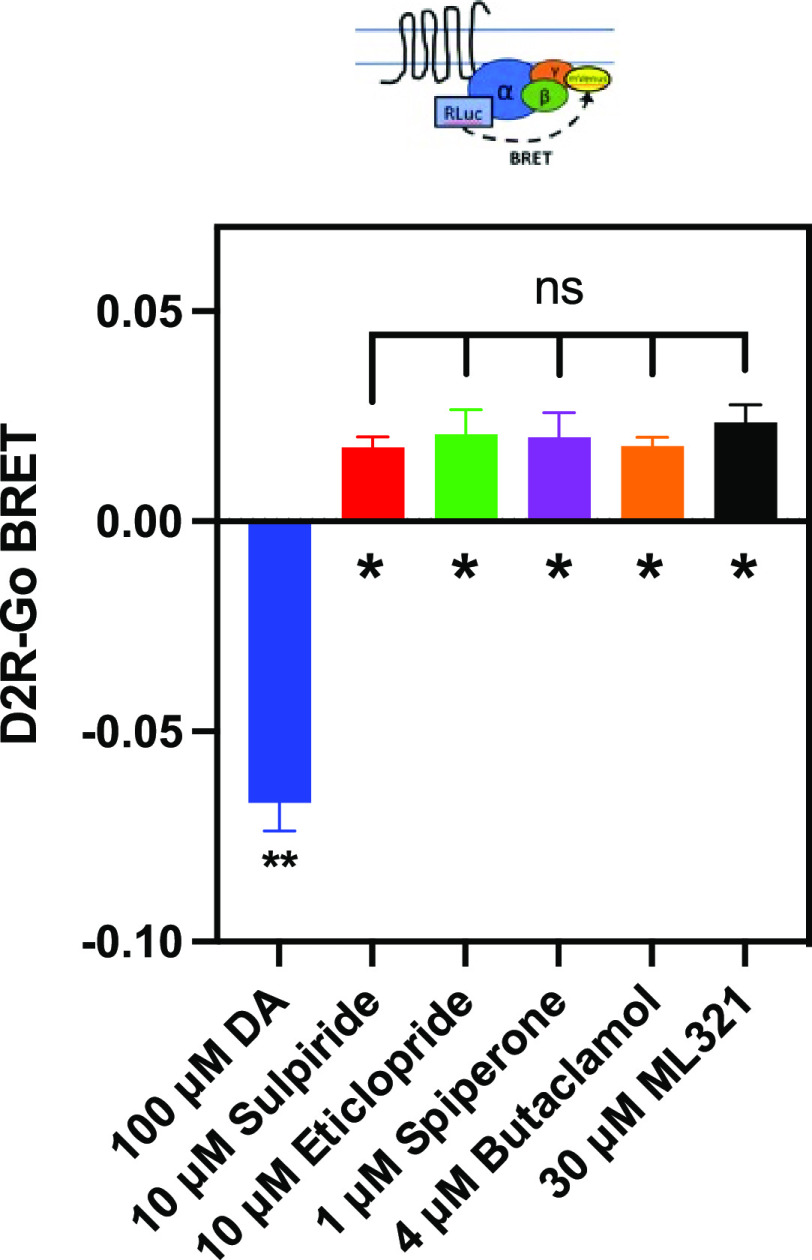
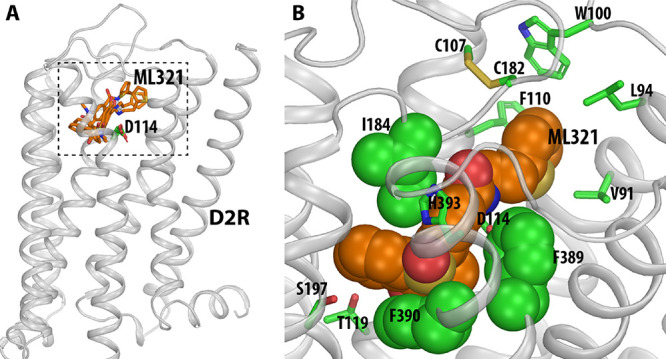
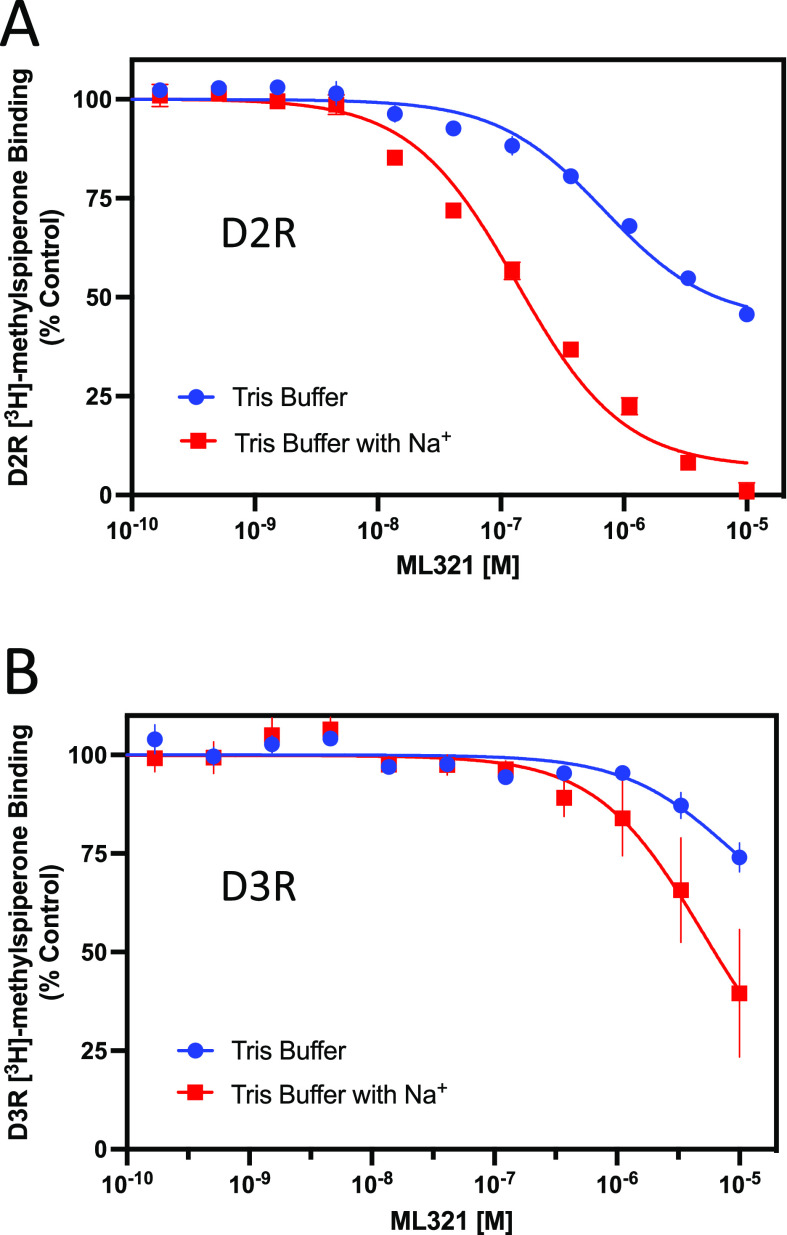
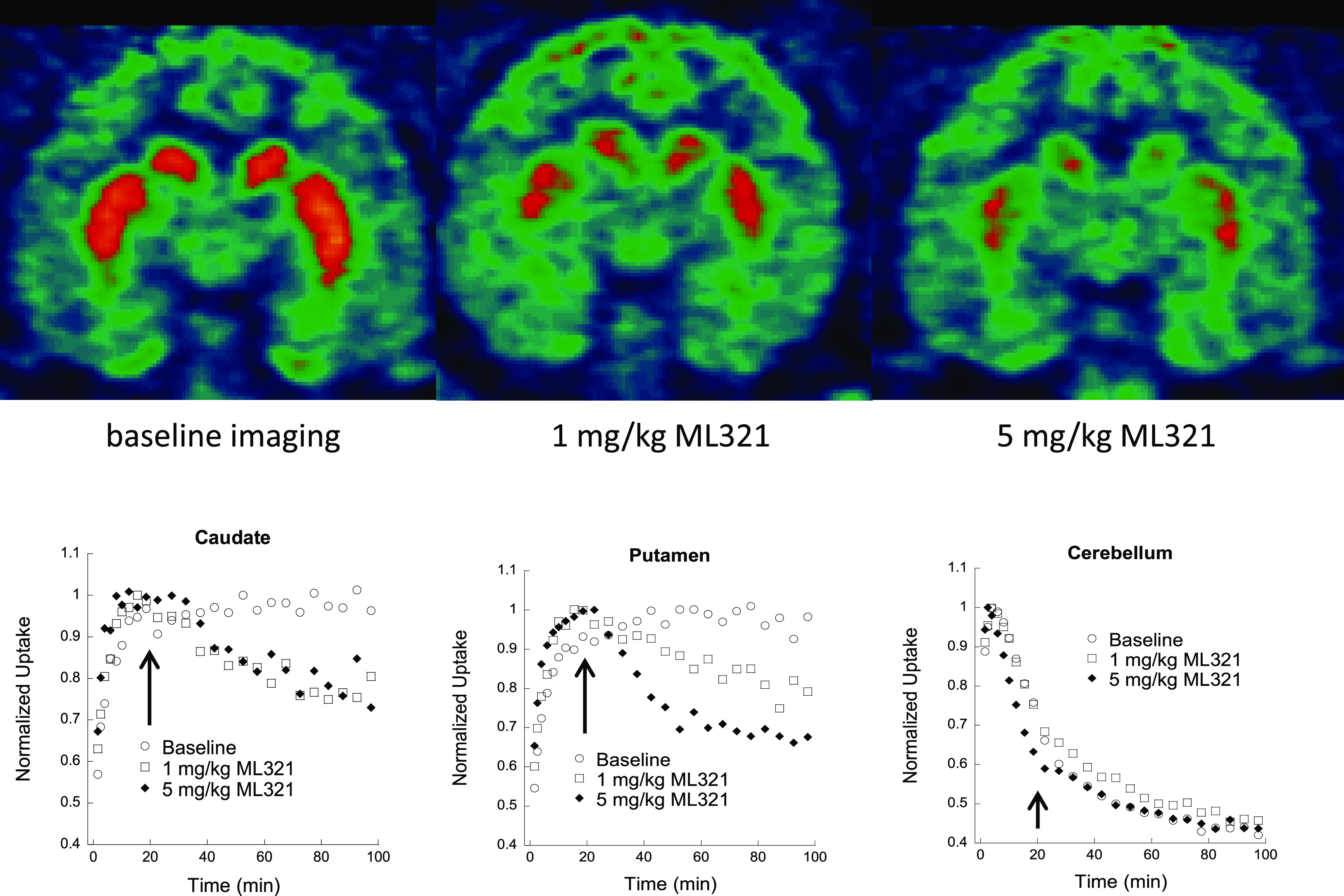
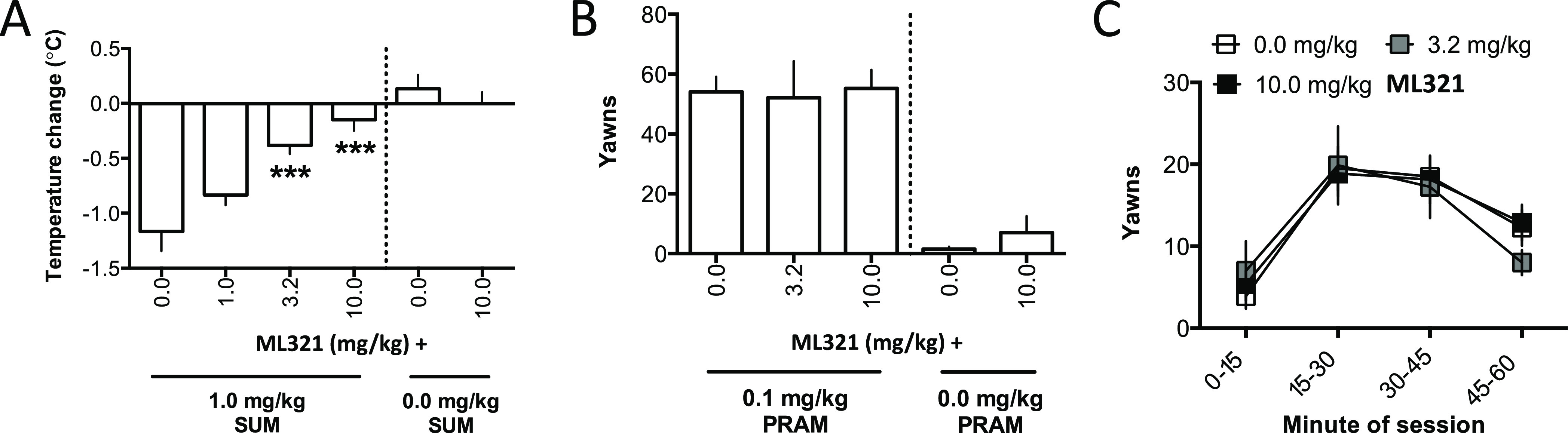
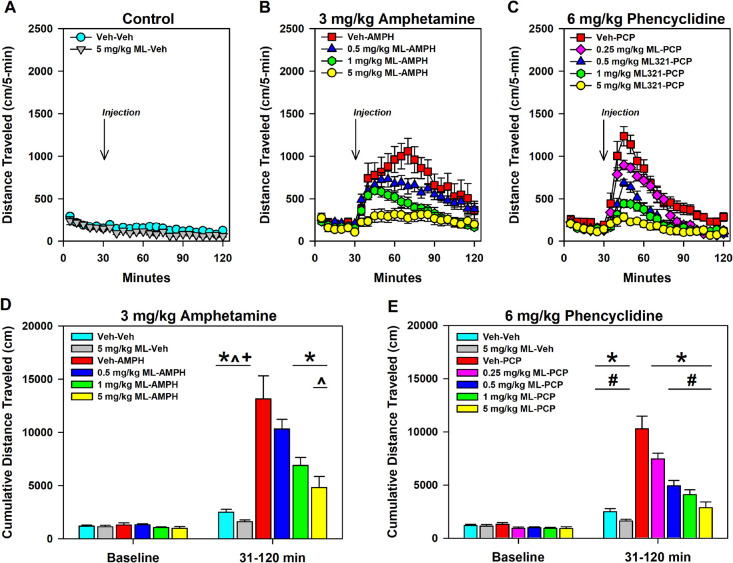
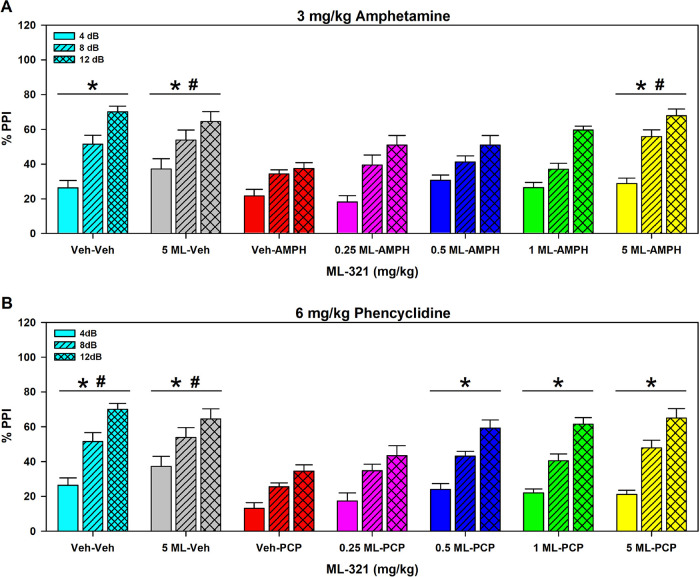
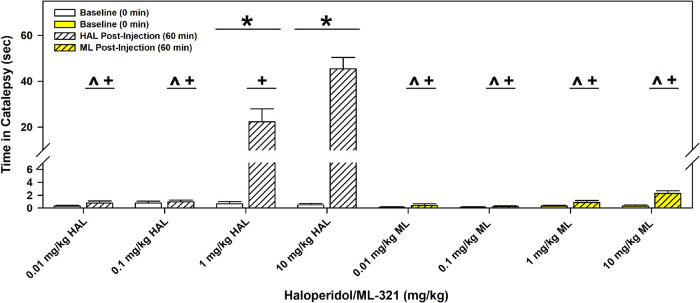
We have developed and characterized a novel D2R antagonist with exceptional GPCR selectivity - ML321. In functional profiling screens of 168 different GPCRs, ML321 showed little activity beyond potent inhibition of the D2R and to a lesser extent the D3R, demonstrating excellent receptor selectivity. The D2R selectivity of ML321 may be related to the fact that, unlike other monoaminergic ligands, ML321 lacks a positively charged amine group and adopts a unique binding pose within the orthosteric binding site of the D2R. PET imaging studies in non-human primates demonstrated that ML321 penetrates the CNS and occupies the D2R in a dose-dependent manner. Behavioral paradigms in rats demonstrate that ML321 can selectively antagonize a D2R-mediated response (hypothermia) while not affecting a D3R-mediated response (yawning) using the same dose of drug, thus indicating exceptional in vivo selectivity. We also investigated the effects of ML321 in animal models that are predictive of antipsychotic efficacy in humans. We found that ML321 attenuates both amphetamine- and phencyclidine-induced locomotor activity and restored pre-pulse inhibition (PPI) of acoustic startle in a dose-dependent manner. Surprisingly, using doses that were maximally effective in both the locomotor and PPI studies, ML321 was relatively ineffective in promoting catalepsy. Kinetic studies revealed that ML321 exhibits slow-on and fast-off receptor binding rates, similar to those observed with atypical antipsychotics with reduced extrapyramidal side effects. Taken together, these observations suggest that ML321, or a derivative thereof, may exhibit ″atypical″ antipsychotic activity in humans with significantly fewer side effects than observed with the currently FDA-approved D2R antagonists.
© 2022 American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources