

Molecular characterization of Richter syndrome identifies de novo diffuse large B-cell lymphomas with poor prognosis
- PMID: 36658118
- PMCID: PMC9852595
- DOI: 10.1038/s41467-022-34642-6
Molecular characterization of Richter syndrome identifies de novo diffuse large B-cell lymphomas with poor prognosis
Abstract
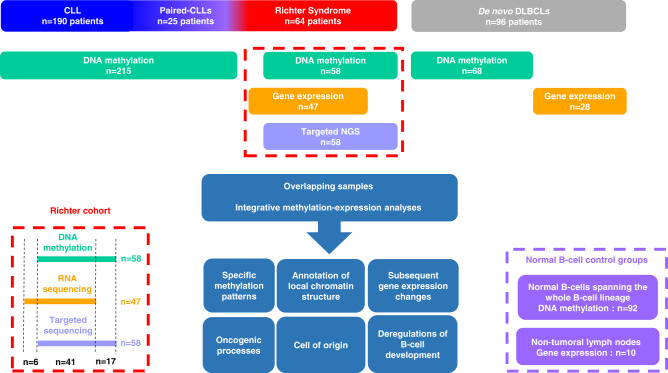
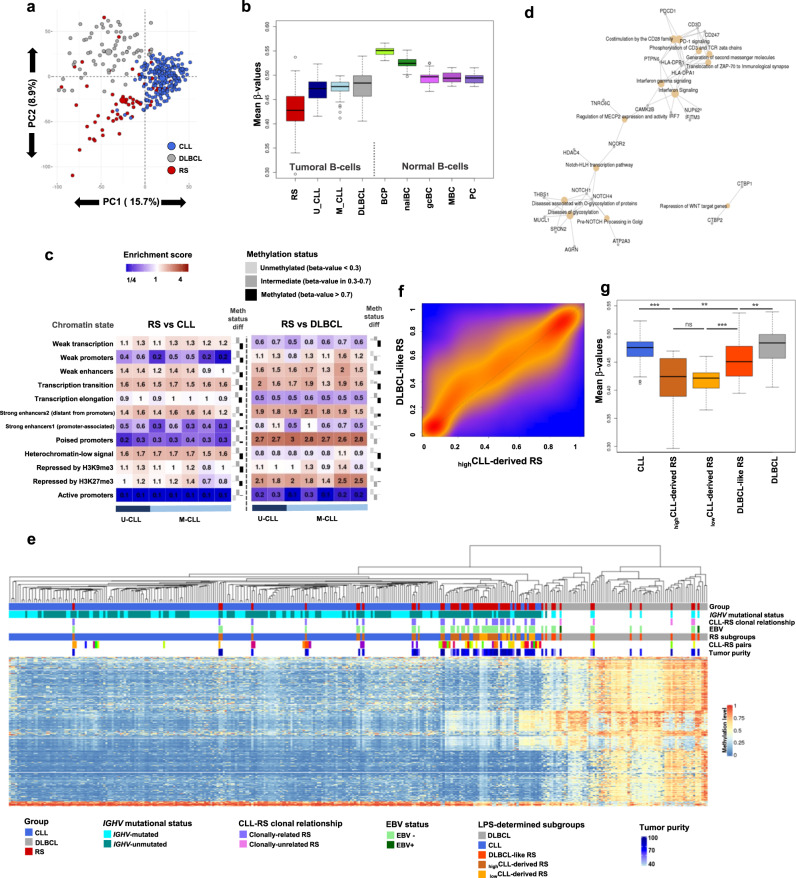
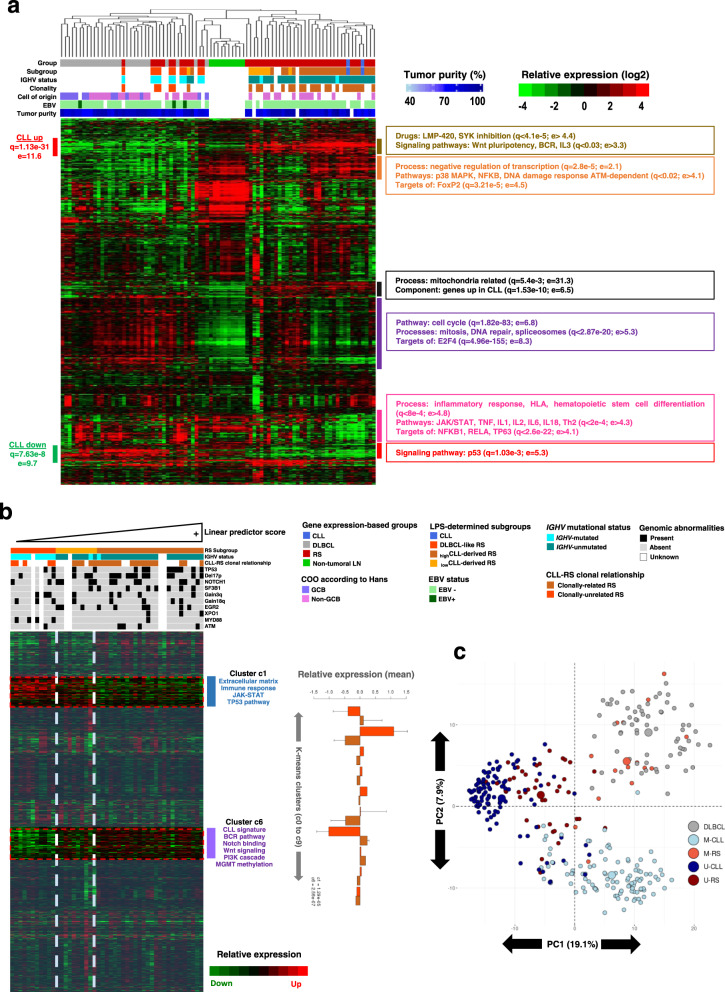
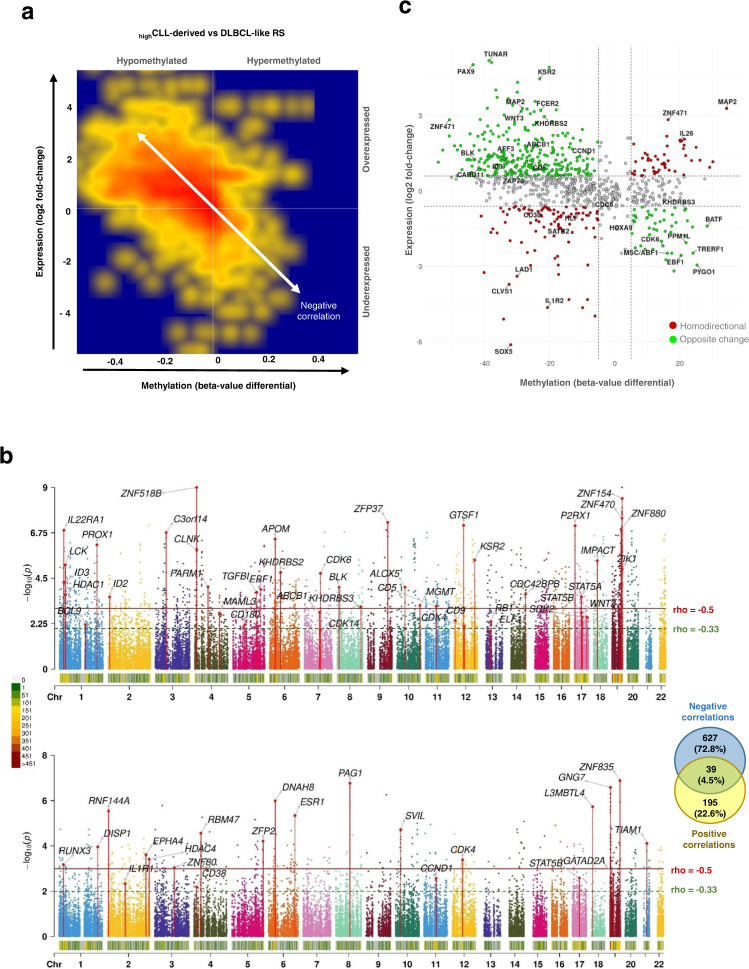
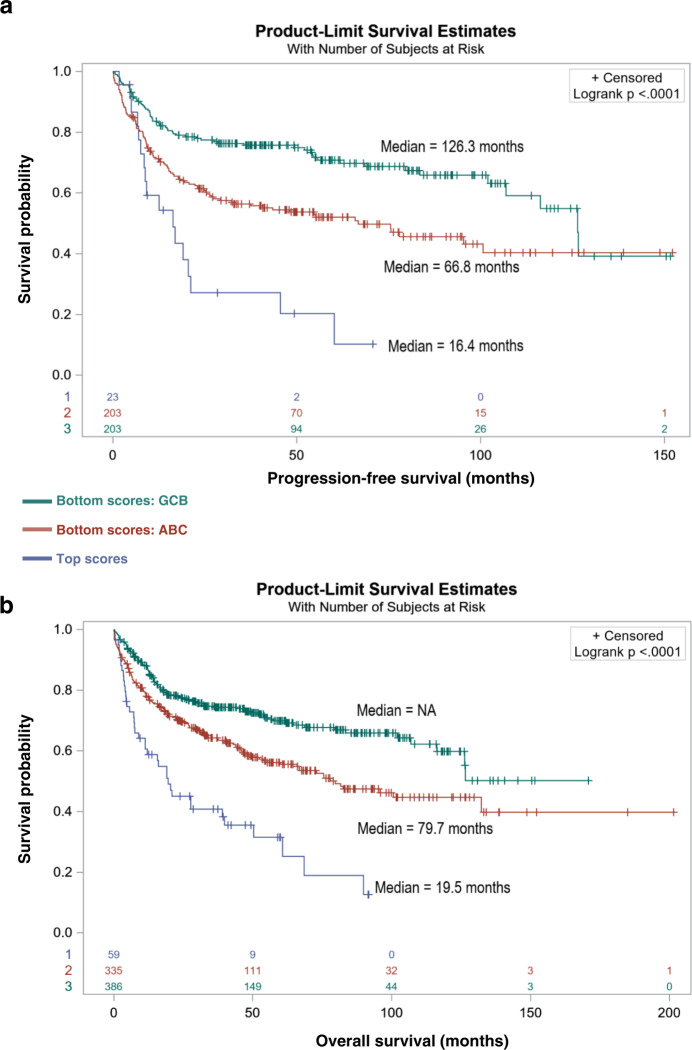
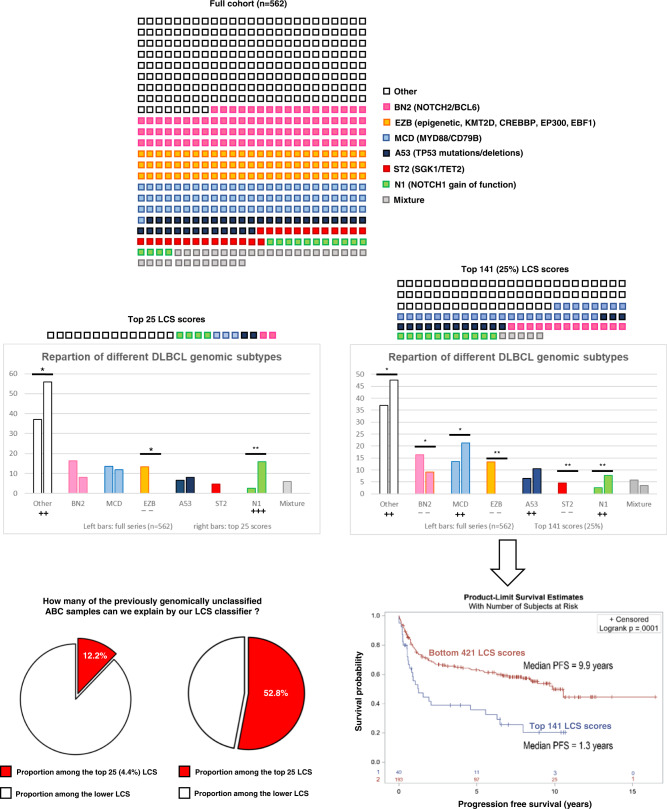
Richter syndrome (RS) is the transformation of chronic lymphocytic leukemia (CLL) into aggressive lymphoma, most commonly diffuse large B-cell lymphoma (DLBCL). We characterize 58 primary human RS samples by genome-wide DNA methylation and whole-transcriptome profiling. Our comprehensive approach determines RS DNA methylation profile and unravels a CLL epigenetic imprint, allowing CLL-RS clonal relationship assessment without the need of the initial CLL tumor DNA. DNA methylation- and transcriptomic-based classifiers were developed, and testing on landmark DLBCL datasets identifies a poor-prognosis, activated B-cell-like DLBCL subset in 111/1772 samples. The classification robustly identifies phenotypes very similar to RS with a specific genomic profile, accounting for 4.3-8.3% of de novo DLBCLs. In this work, RS multi-omics characterization determines oncogenic mechanisms, establishes a surrogate marker for CLL-RS clonal relationship, and provides a clinically relevant classifier for a subset of primary "RS-type DLBCL" with unfavorable prognosis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Mao Z, et al. IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am. J. Surg. Pathol. 2007;31:1605–1614. doi: 10.1097/PAS.0b013e31804bdaf8. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
