

Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing
- PMID: 36658122
- PMCID: PMC9852470
- DOI: 10.1038/s41467-023-35858-w
Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing
Abstract
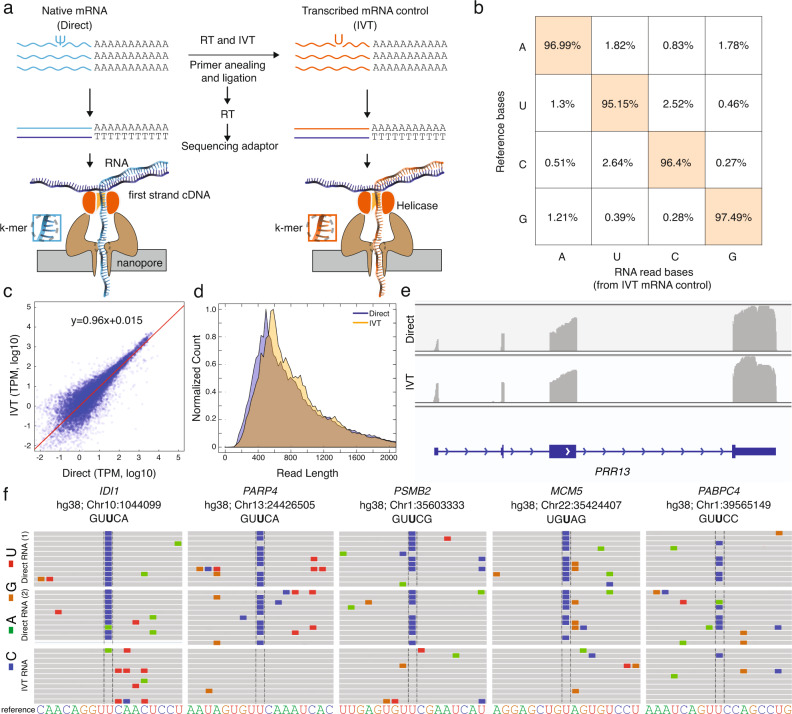
Here, we develop and apply a semi-quantitative method for the high-confidence identification of pseudouridylated sites on mammalian mRNAs via direct long-read nanopore sequencing. A comparative analysis of a modification-free transcriptome reveals that the depth of coverage and specific k-mer sequences are critical parameters for accurate basecalling. By adjusting these parameters for high-confidence U-to-C basecalling errors, we identify many known sites of pseudouridylation and uncover previously unreported uridine-modified sites, many of which fall in k-mers that are known targets of pseudouridine synthases. Identified sites are validated using 1000-mer synthetic RNA controls bearing a single pseudouridine in the center position, demonstrating systematic under-calling using our approach. We identify mRNAs with up to 7 unique modification sites. Our workflow allows direct detection of low-, medium-, and high-occupancy pseudouridine modifications on native RNA molecules from nanopore sequencing data and multiple modifications on the same strand.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
