

The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma
- PMID: 36658220
- PMCID: PMC9928584
- DOI: 10.1038/s41556-022-01059-8
The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma
Abstract
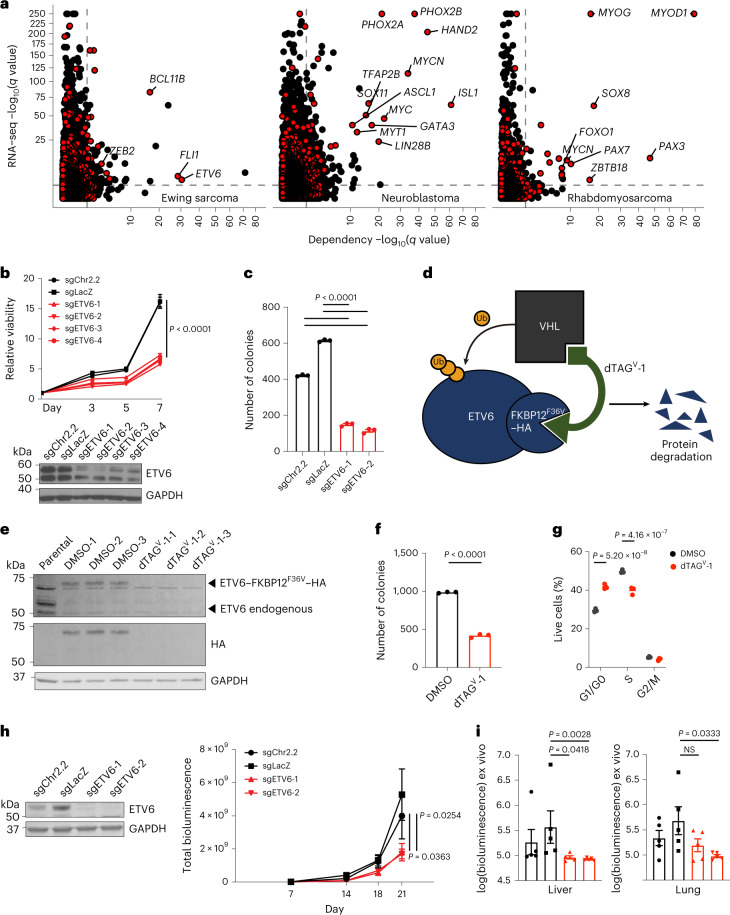
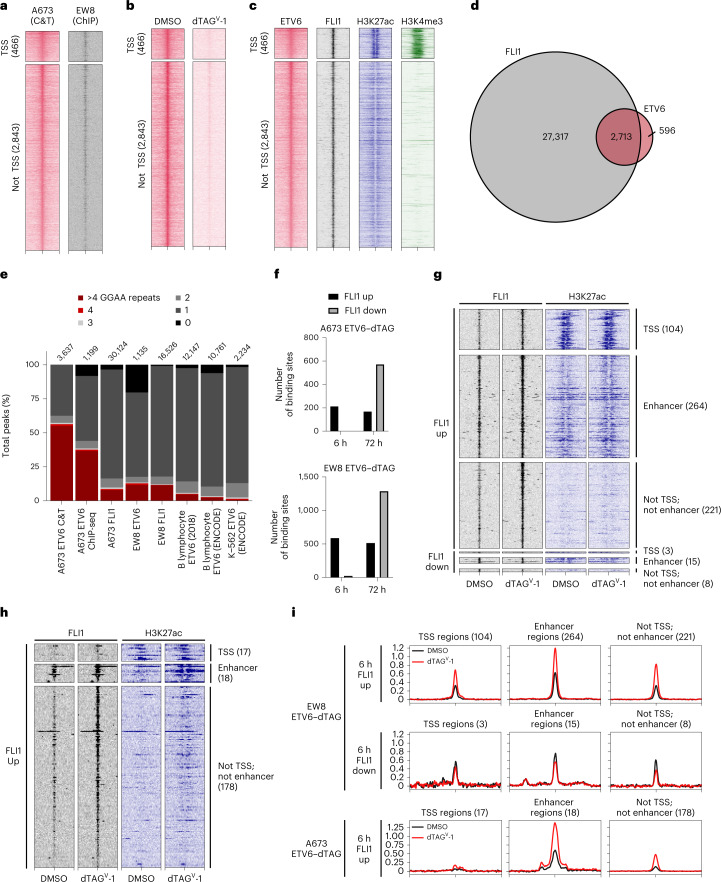
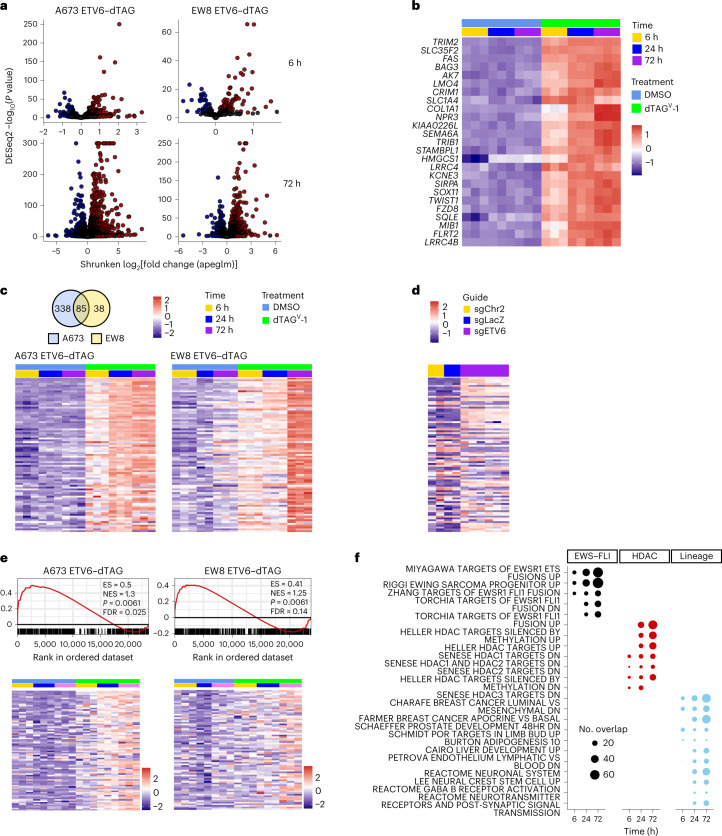
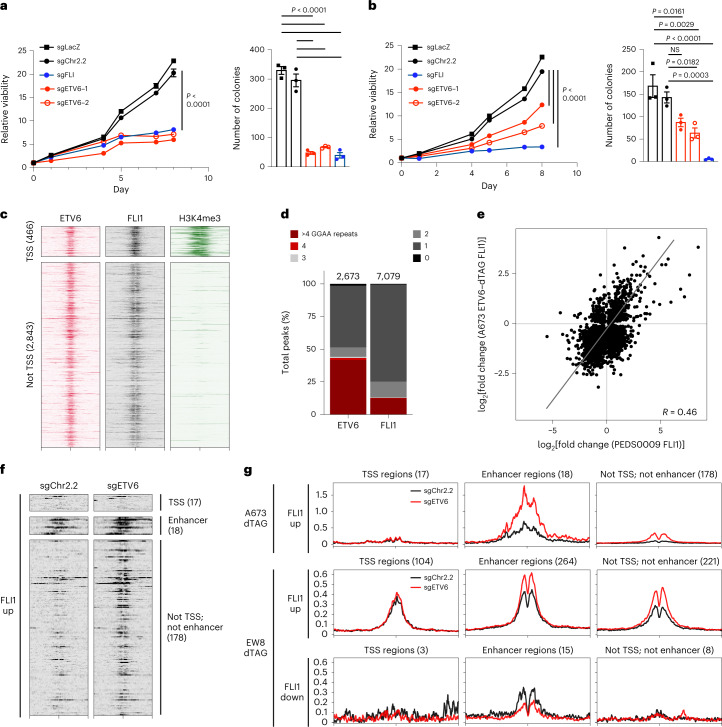
Transcription factors (TFs) are frequently mutated in cancer. Paediatric cancers exhibit few mutations genome-wide but frequently harbour sentinel mutations that affect TFs, which provides a context to precisely study the transcriptional circuits that support mutant TF-driven oncogenesis. A broadly relevant mechanism that has garnered intense focus involves the ability of mutant TFs to hijack wild-type lineage-specific TFs in self-reinforcing transcriptional circuits. However, it is not known whether this specific type of circuitry is equally crucial in all mutant TF-driven cancers. Here we describe an alternative yet central transcriptional mechanism that promotes Ewing sarcoma, wherein constraint, rather than reinforcement, of the activity of the fusion TF EWS-FLI supports cancer growth. We discover that ETV6 is a crucial TF dependency that is specific to this disease because it, counter-intuitively, represses the transcriptional output of EWS-FLI. This work discovers a previously undescribed transcriptional mechanism that promotes cancer.
© 2023. The Author(s).
Conflict of interest statement
K.S. receives grant funding from the DFCI/Novartis Drug Discovery Program and from KronosBio, is a member of the SAB and has stock options with Auron Therapeutics and has consulted for AstraZeneca. S.L.L. declares a competing interest as a member of the advisory board for Salarius Pharmaceuticals and is a listed the inventor on US patent no. US 7,939,253 B2, ‘Methods and compositions for the diagnosis and treatment of Ewing’s sarcoma’ and US patent no. US 8,557,532, ‘Diagnosis and treatment of drug-resistant Ewing’s sarcoma.’ N.V.D. is an employee of Genentech, a member of the Roche Group. N.S.G. is a founder, scientific advisory board member and equity holder in Syros, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member) and Soltego (board member). The remaining authors declare no competing interests.
Figures

Comment in
-
Oncogenic role for an EWS-FLI1 suppressor.Nat Cell Biol. 2023 Feb;25(2):214-216. doi: 10.1038/s41556-022-01067-8. Nat Cell Biol. 2023. PMID: 36658218 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
