

Genome-wide RNA polymerase stalling shapes the transcriptome during aging
- PMID: 36658433
- PMCID: PMC9925383
- DOI: 10.1038/s41588-022-01279-6
Genome-wide RNA polymerase stalling shapes the transcriptome during aging
Abstract
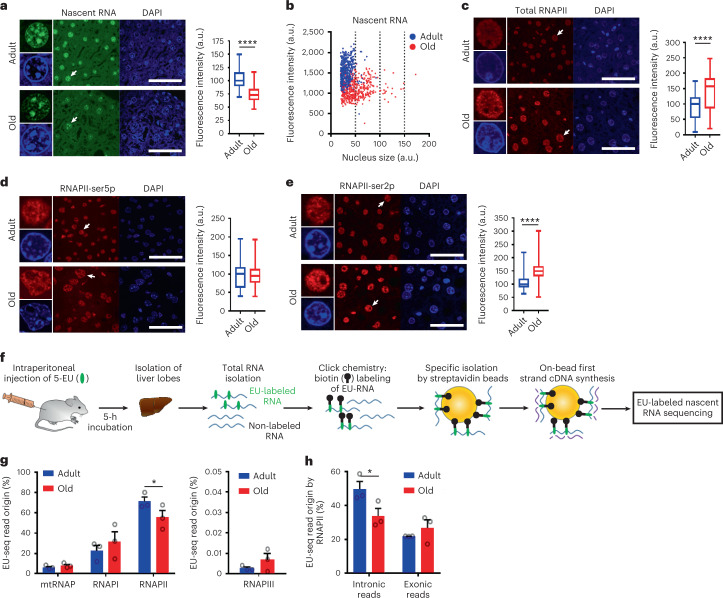
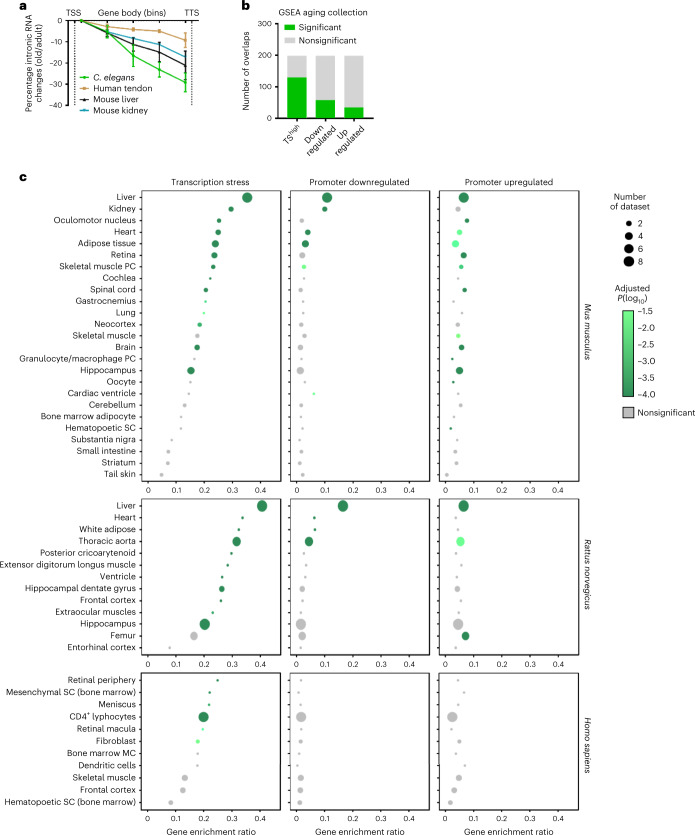
Gene expression profiling has identified numerous processes altered in aging, but how these changes arise is largely unknown. Here we combined nascent RNA sequencing and RNA polymerase II chromatin immunoprecipitation followed by sequencing to elucidate the underlying mechanisms triggering gene expression changes in wild-type aged mice. We found that in 2-year-old liver, 40% of elongating RNA polymerases are stalled, lowering productive transcription and skewing transcriptional output in a gene-length-dependent fashion. We demonstrate that this transcriptional stress is caused by endogenous DNA damage and explains the majority of gene expression changes in aging in most mainly postmitotic organs, specifically affecting aging hallmark pathways such as nutrient sensing, autophagy, proteostasis, energy metabolism, immune function and cellular stress resilience. Age-related transcriptional stress is evolutionary conserved from nematodes to humans. Thus, accumulation of stochastic endogenous DNA damage during aging deteriorates basal transcription, which establishes the age-related transcriptome and causes dysfunction of key aging hallmark pathways, disclosing how DNA damage functionally underlies major aspects of normal aging.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
