

Notch1 mutations drive clonal expansion in normal esophageal epithelium but impair tumor growth
- PMID: 36658434
- PMCID: PMC9925379
- DOI: 10.1038/s41588-022-01280-z
Notch1 mutations drive clonal expansion in normal esophageal epithelium but impair tumor growth
Abstract
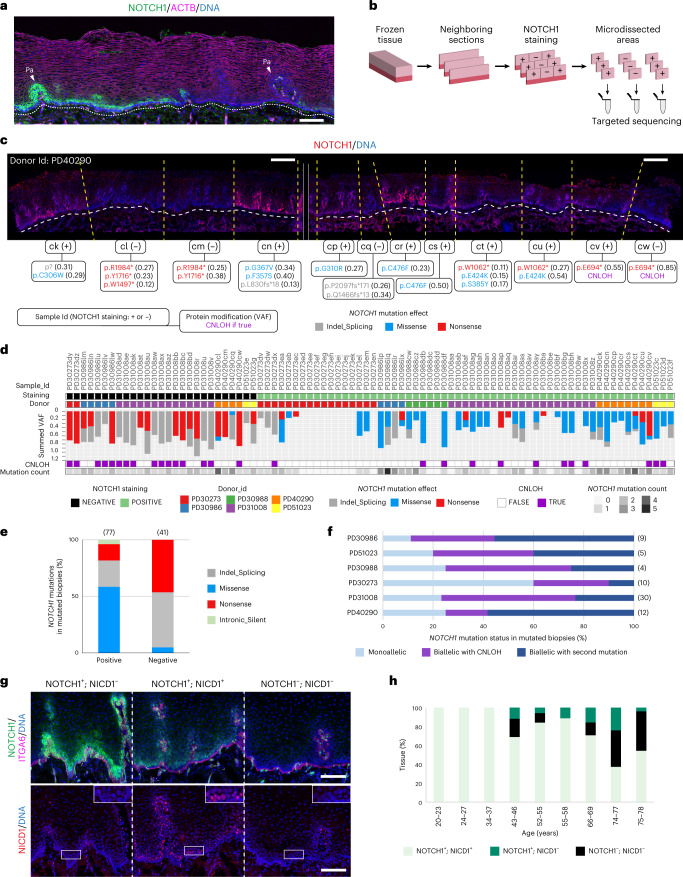
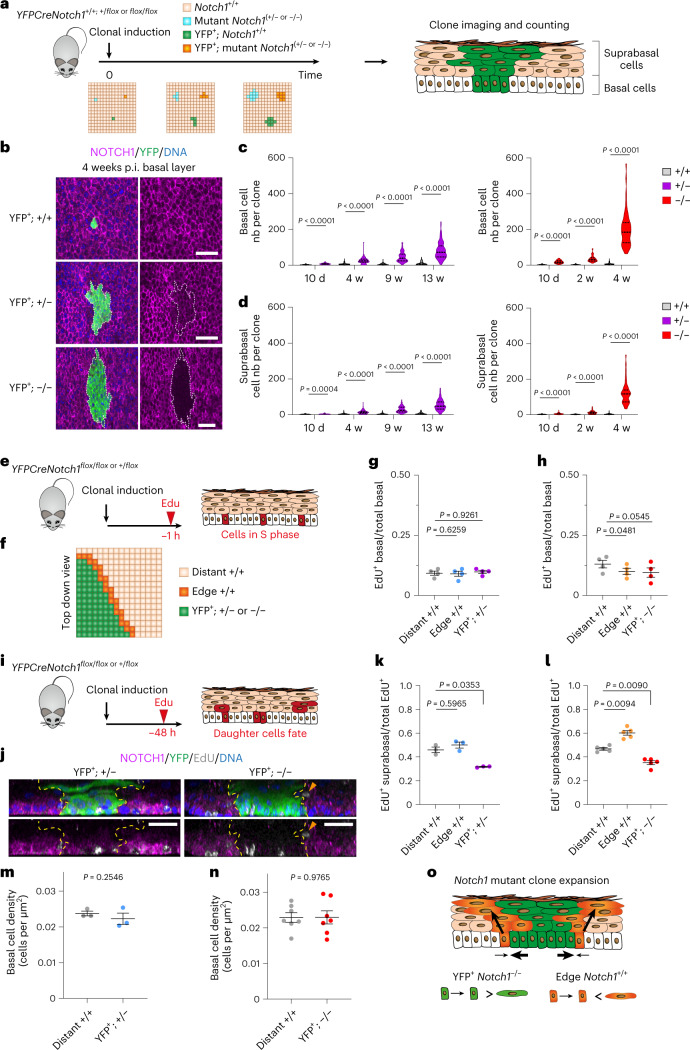
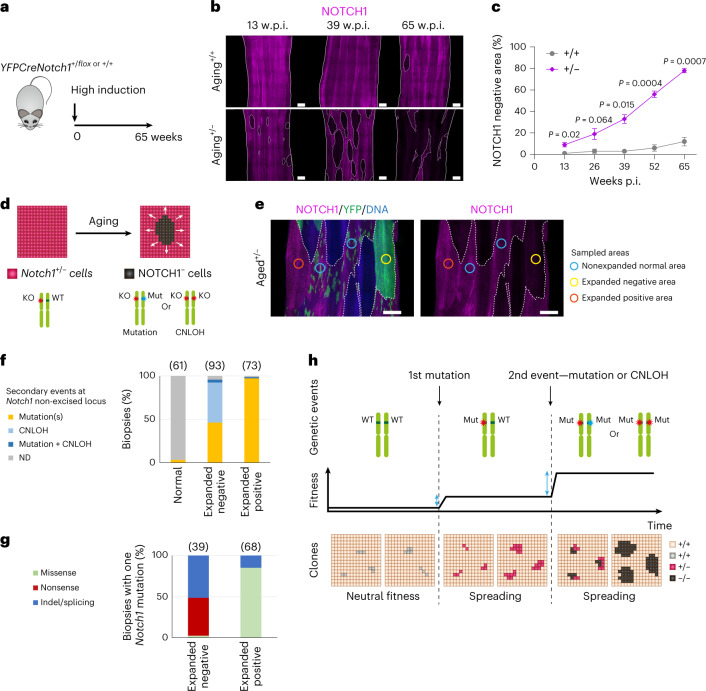
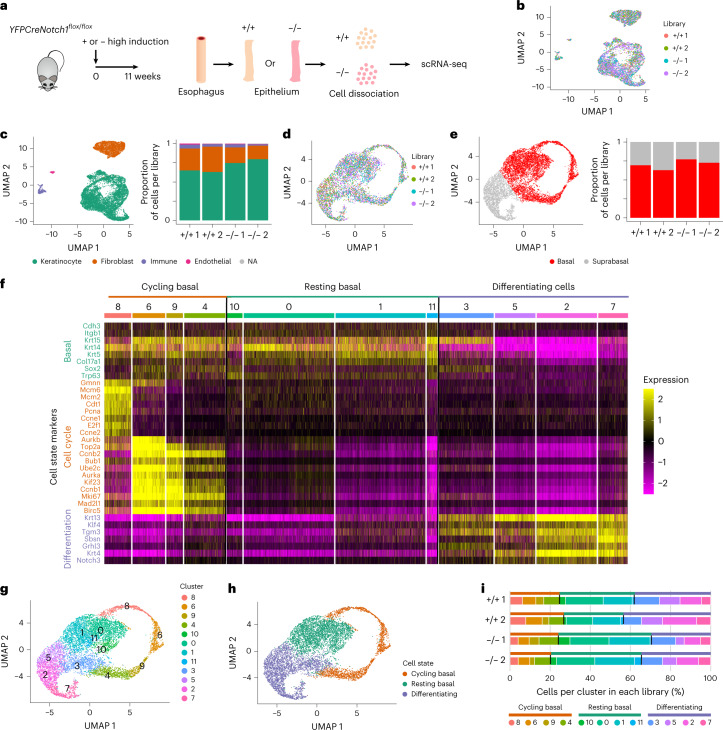
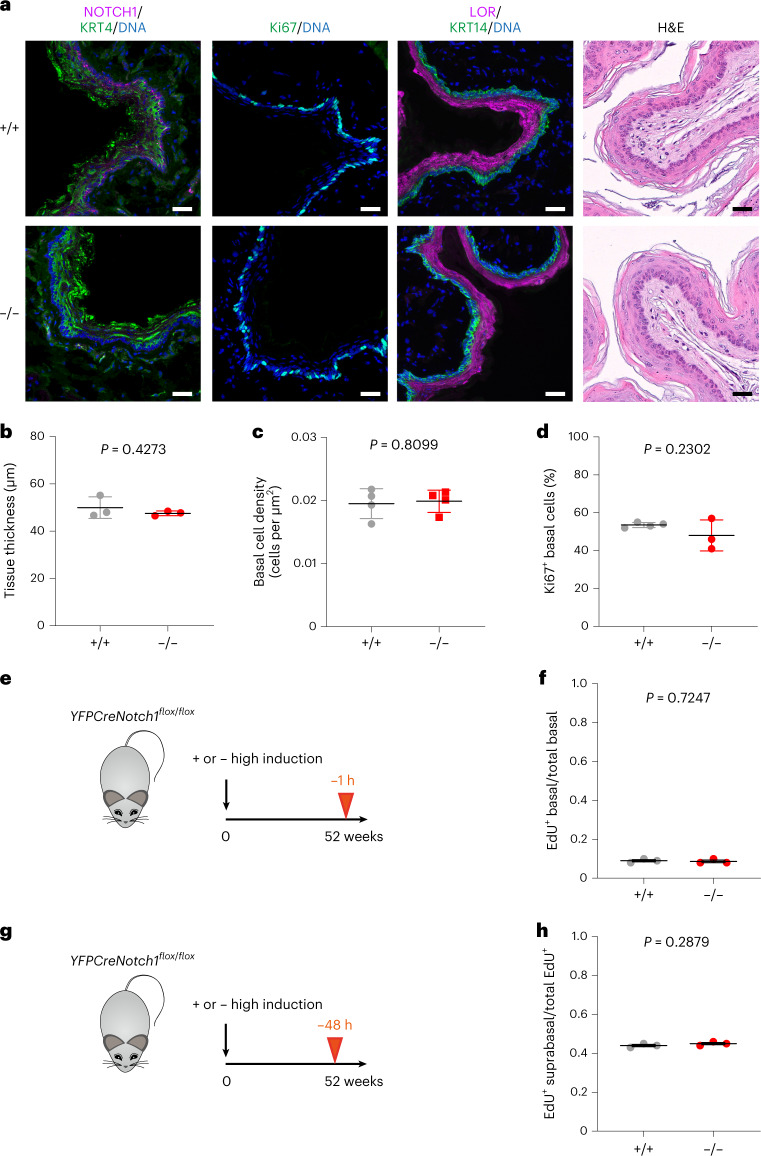
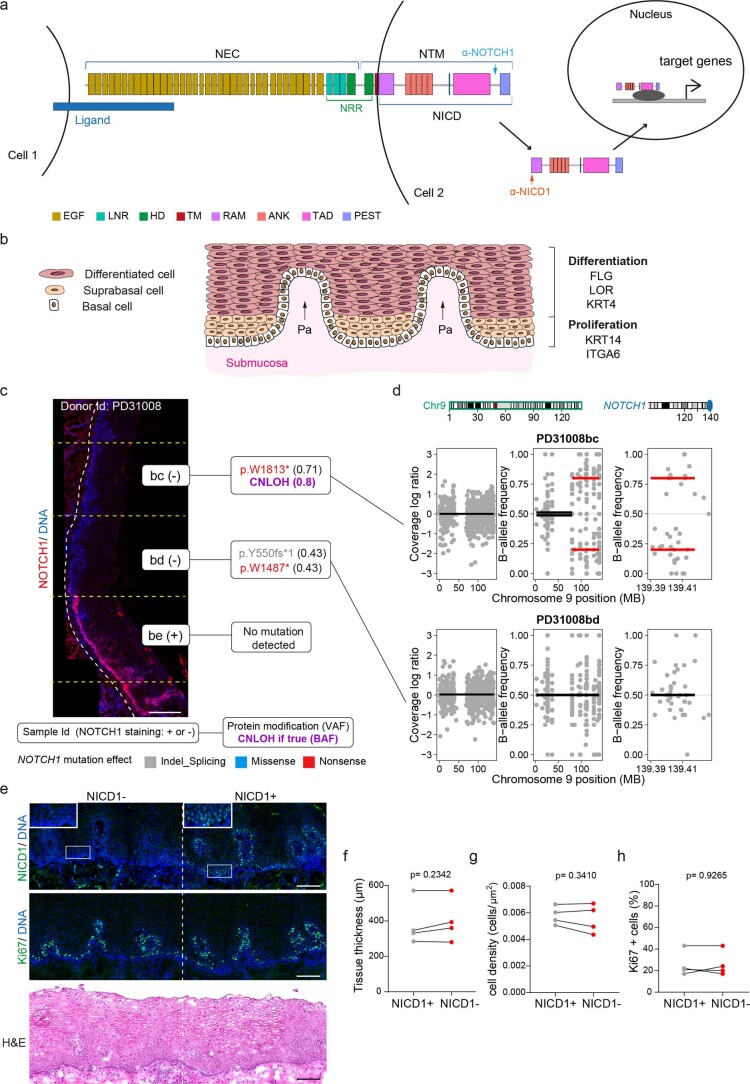
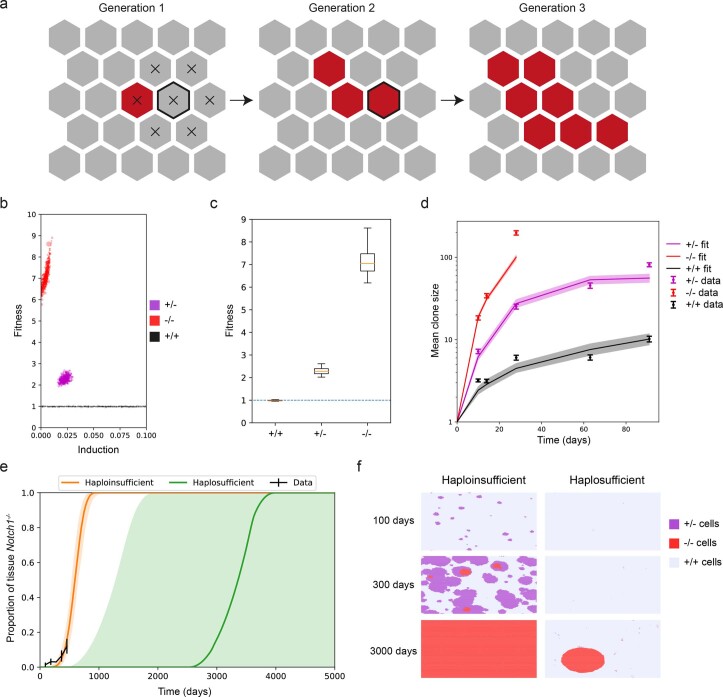
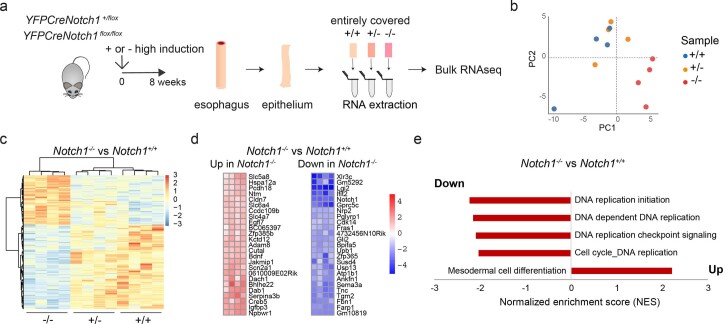
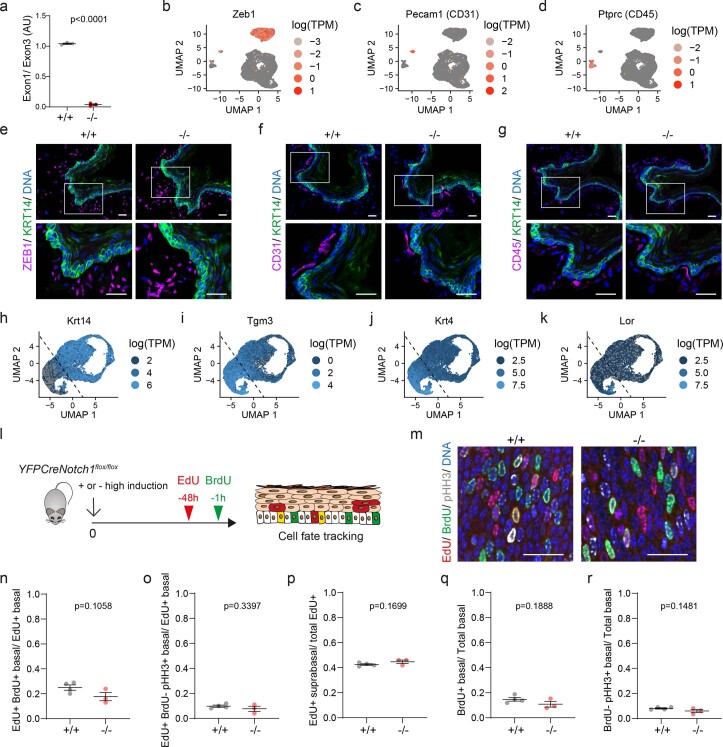
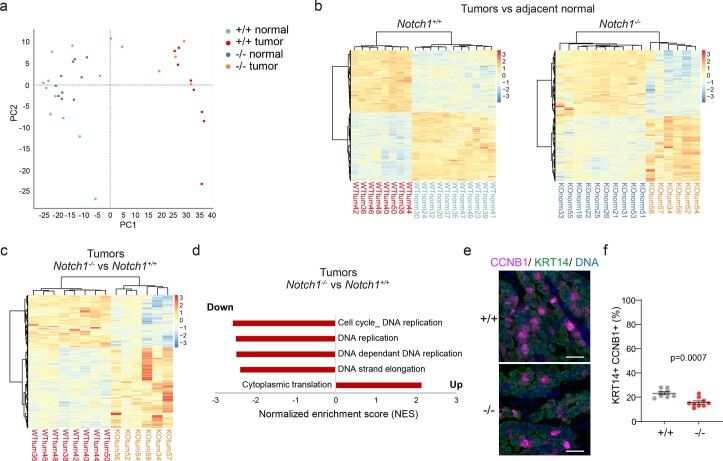
NOTCH1 mutant clones occupy the majority of normal human esophagus by middle age but are comparatively rare in esophageal cancers, suggesting NOTCH1 mutations drive clonal expansion but impede carcinogenesis. Here we test this hypothesis. Sequencing NOTCH1 mutant clones in aging human esophagus reveals frequent biallelic mutations that block NOTCH1 signaling. In mouse esophagus, heterozygous Notch1 mutation confers a competitive advantage over wild-type cells, an effect enhanced by loss of the second allele. Widespread Notch1 loss alters transcription but has minimal effects on the epithelial structure and cell dynamics. In a carcinogenesis model, Notch1 mutations were less prevalent in tumors than normal epithelium. Deletion of Notch1 reduced tumor growth, an effect recapitulated by anti-NOTCH1 antibody treatment. Notch1 null tumors showed reduced proliferation. We conclude that Notch1 mutations in normal epithelium are beneficial as wild-type Notch1 favors tumor expansion. NOTCH1 blockade may have therapeutic potential in preventing esophageal squamous cancer.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
