

The B domain of protein A retains residual structures in 6 M guanidinium chloride as revealed by hydrogen/deuterium-exchange NMR spectroscopy
- PMID: 36659853
- PMCID: PMC9926473
- DOI: 10.1002/pro.4569
The B domain of protein A retains residual structures in 6 M guanidinium chloride as revealed by hydrogen/deuterium-exchange NMR spectroscopy
Erratum in
-
CORRECTION.Protein Sci. 2023 Jul;32(7):e4696. doi: 10.1002/pro.4696. Protein Sci. 2023. PMID: 37382354 Free PMC article. No abstract available.
Abstract
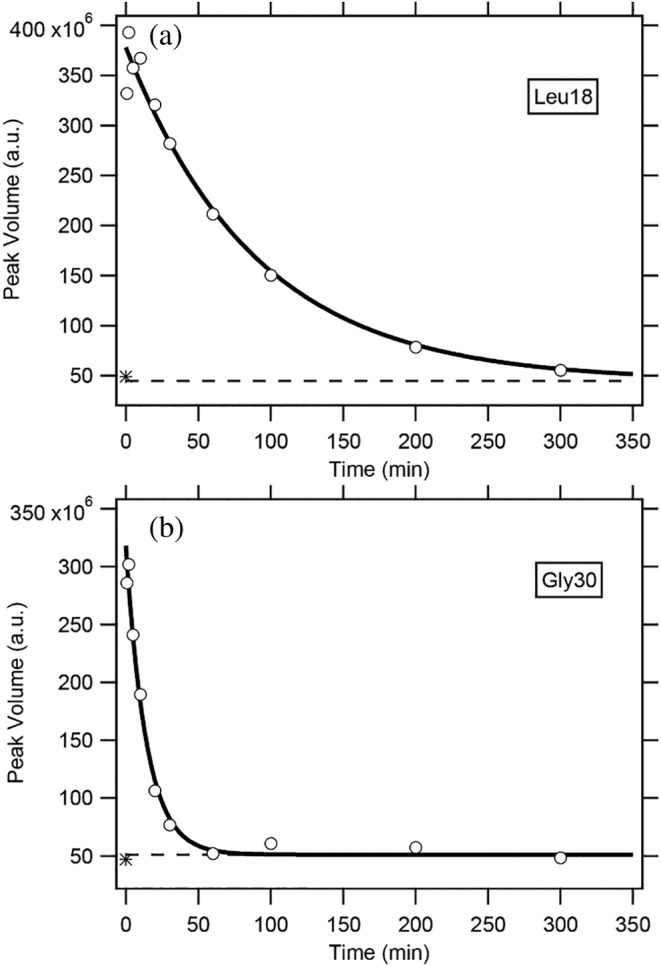
The characterization of residual structures persistent in unfolded proteins is an important issue in studies of protein folding, because the residual structures present, if any, may form a folding initiation site and guide the subsequent folding reactions. Here, we studied the residual structures of the isolated B domain (BDPA) of staphylococcal protein A in 6 M guanidinium chloride. BDPA is a small three-helix-bundle protein, and until recently its folding/unfolding reaction has been treated as a simple two-state process between the native and the fully unfolded states. We employed a dimethylsulfoxide (DMSO)-quenched hydrogen/deuterium (H/D)-exchange 2D NMR techniques with the use of spin desalting columns, which allowed us to investigate the H/D-exchange behavior of individually identified peptide amide (NH) protons. We obtained H/D-exchange protection factors of the 21 NH protons that form an α-helical hydrogen bond in the native structure, and the majority of these NH protons were significantly protected with a protection factor of 2.0-5.2 in 6 M guanidinium chloride, strongly suggesting that these weakly protected NH protons form much stronger hydrogen bonds under native folding conditions. The results can be used to deduce the structure of an early folding intermediate, when such an intermediate is shown by other methods. Among three native helical regions, the third helix in the C-terminal side was highly protected and stabilized by side-chain salt bridges, probably acting as the folding initiation site of BDPA. The present results are discussed in relation to previous experimental and computational findings on the folding mechanisms of BDPA.
Keywords: 2D NMR; hydrogen/deuterium exchange; protein folding; residual structure; unfolded state.
© 2023 The Authors. Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
