

VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases
- PMID: 36662445
- PMCID: PMC10082120
- DOI: 10.1007/s11739-023-03193-z
VEXAS syndrome: a new paradigm for adult-onset monogenic autoinflammatory diseases
Abstract
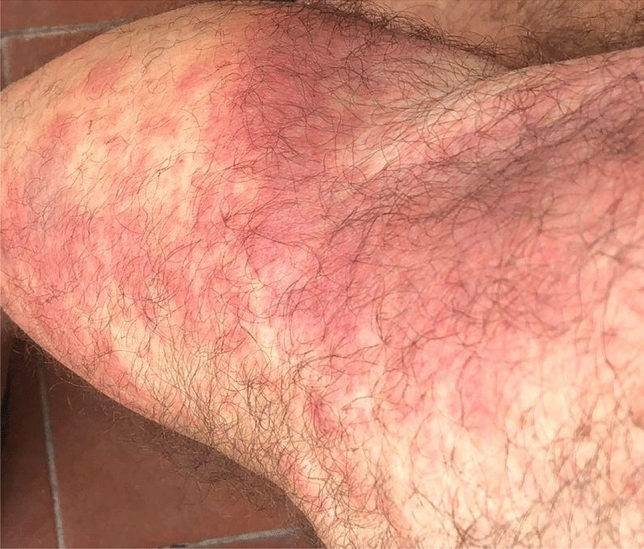
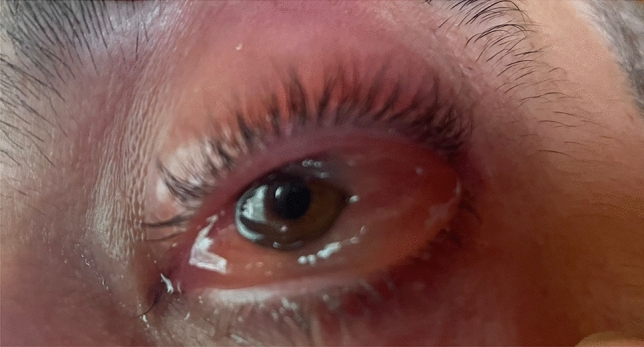
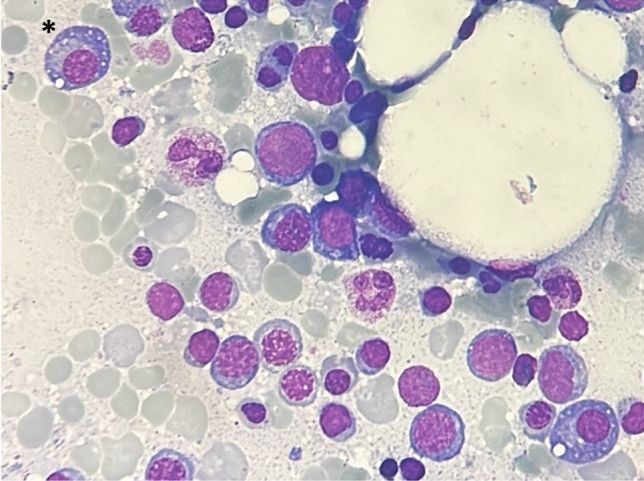
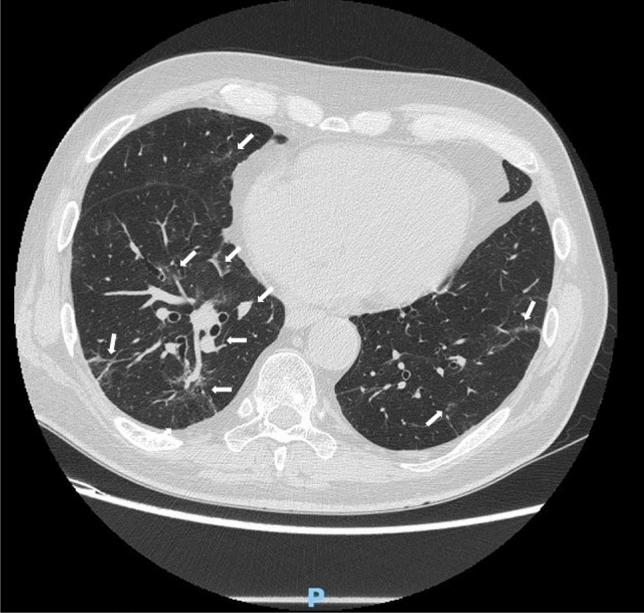
VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) syndrome is a recently described pathological entity. It is an acquired monogenic autoinflammatory disease caused by somatic mutations of the UBA1 gene in blood cells precursors; the gene encodes one of the two E1 enzyme isoforms that initiates ubiquitylation in cell's cytoplasm. VEXAS syndrome leads to systemic inflammation, with all organs and tissues potentially involved. The clinical picture may be extremely heterogenous, mimicking different other systemic rheumatologic entities coexisting with haematological disorders, especially myelodysplastic syndrome. This new disease represents a very intriguing clinical condition in several respects: it accounts for the paradigm of adult-onset monogenic autoinflammatory diseases determined by a genetic mosaicism resulting in the development of a challenging multiorgan inflammatory condition. Moreover, VEXAS syndrome is perhaps not an exceptionally rare condition and represents an example of a systemic genetic autoinflammatory disease drawing its origin in bone marrow disorders. VEXAS syndrome should be strongly considered in each adult patient with an unexplained systemic inflammatory condition, especially when recurrent fevers, neutrophilic dermatosis, relapsing polychondritis, ocular inflammation and other systemic inflammatory symptoms accompanying myelodysplastic syndrome or other haematological disorders. The syndrome deserves a multidisciplinary approach to reach the diagnosis and ensure the best management of a potentially very challenging condition. To quickly describe the clinical course, long-term outcomes, and the optimal management of this new syndrome it is essential to join forces internationally. To this end, the international AutoInflammatory Disease Alliance (AIDA) registry dedicated to VEXAS syndrome has been developed and is already active.
Keywords: Diagnosis; Genetics; Haematology; Monogenic autoinflammatory diseases; Ocular inflammation; Treatment.
© 2023. The Author(s).
Conflict of interest statement
The author(s) declare that they have no conflict of interest.
Figures


References
-
- Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol. 2022;186:564–574. doi: 10.1111/bjd.20805. - DOI - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
