

SWI/SNF Blockade Disrupts PU.1-Directed Enhancer Programs in Normal Hematopoietic Cells and Acute Myeloid Leukemia
- PMID: 36662812
- PMCID: PMC10071820
- DOI: 10.1158/0008-5472.CAN-22-2129
SWI/SNF Blockade Disrupts PU.1-Directed Enhancer Programs in Normal Hematopoietic Cells and Acute Myeloid Leukemia
Abstract
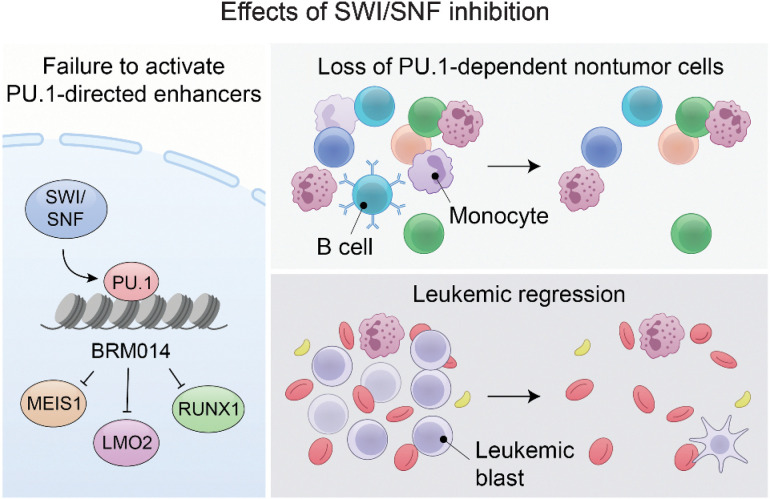
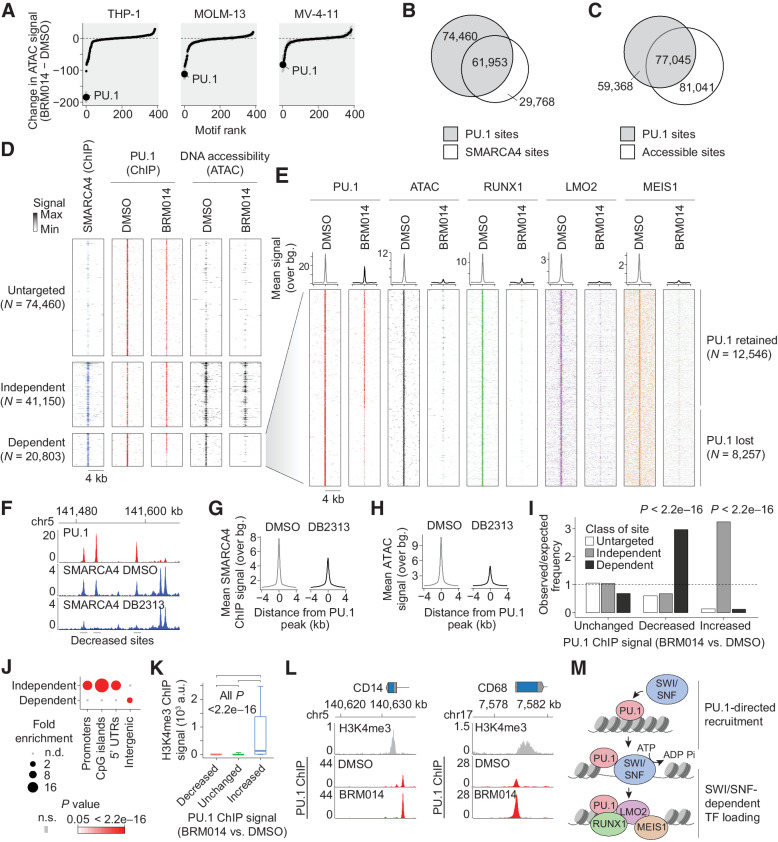
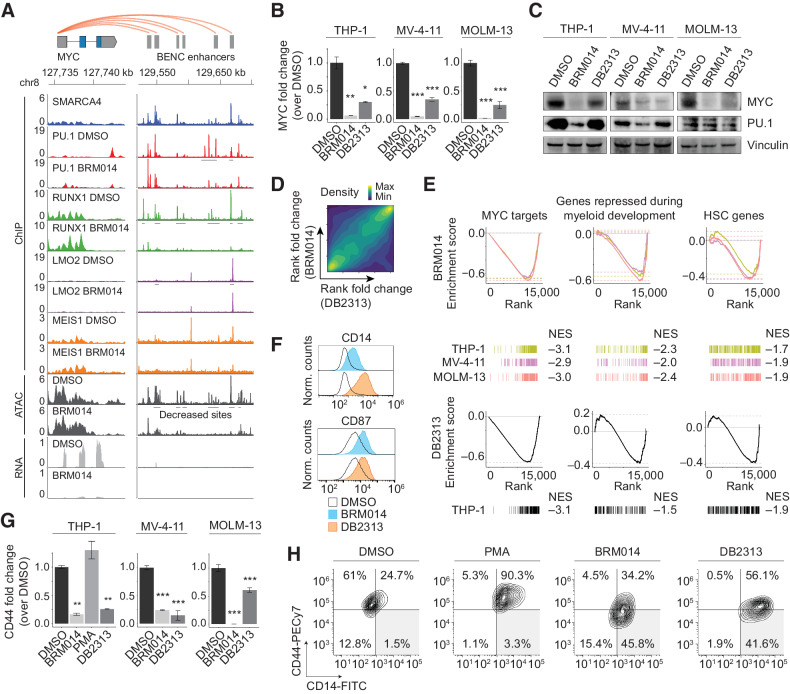
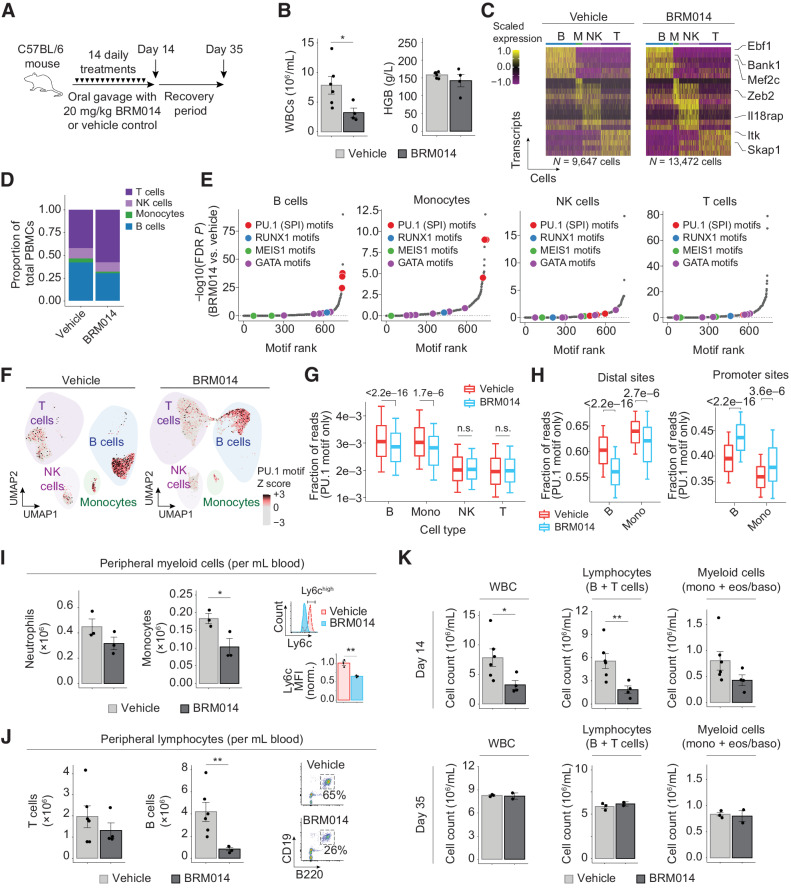
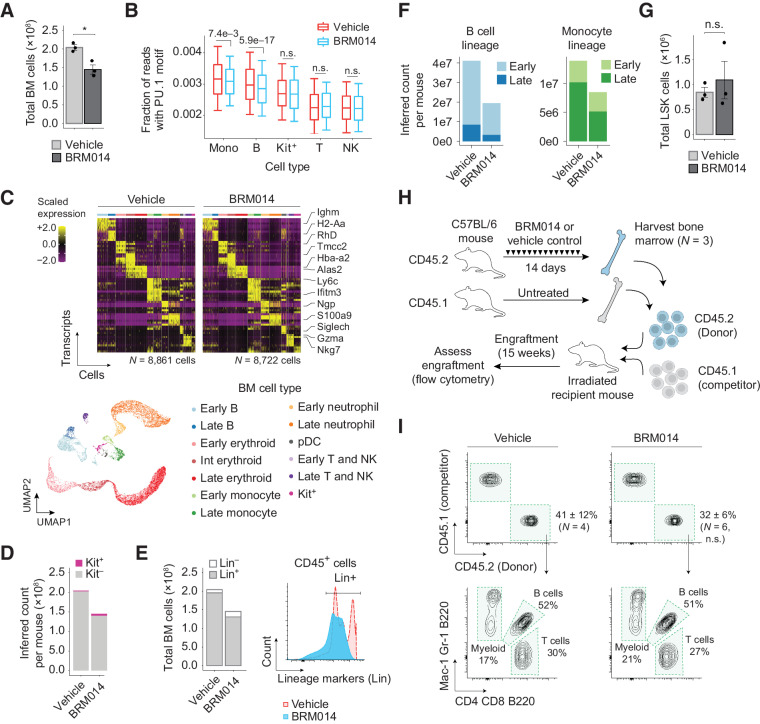
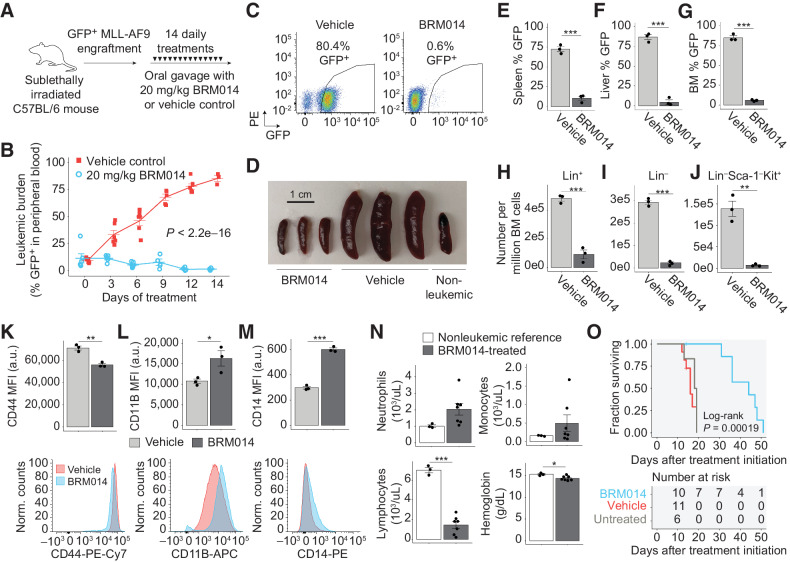
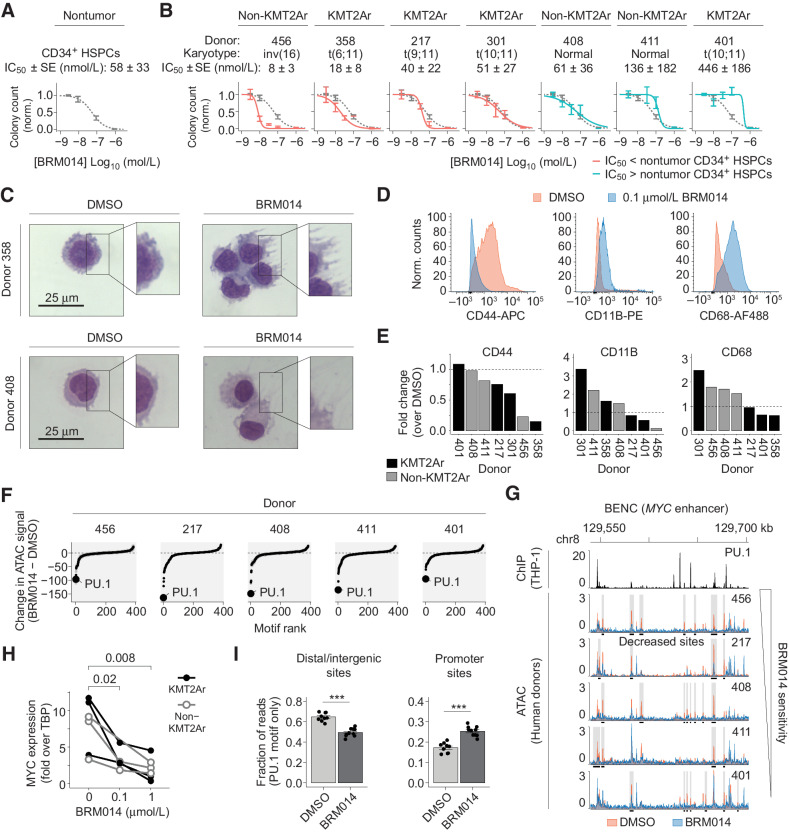
In acute myeloid leukemia (AML), SWI/SNF chromatin remodeling complexes sustain leukemic identity by driving high levels of MYC. Previous studies have implicated the hematopoietic transcription factor PU.1 (SPI1) as an important target of SWI/SNF inhibition, but PU.1 is widely regarded to have pioneer-like activity. As a result, many questions have remained regarding the interplay between PU.1 and SWI/SNF in AML as well as normal hematopoiesis. Here we found that PU.1 binds to most of its targets in a SWI/SNF-independent manner and recruits SWI/SNF to promote accessibility for other AML core regulatory factors, including RUNX1, LMO2, and MEIS1. SWI/SNF inhibition in AML cells reduced DNA accessibility and binding of these factors at PU.1 sites and redistributed PU.1 to promoters. Analysis of nontumor hematopoietic cells revealed that similar effects also impair PU.1-dependent B-cell and monocyte populations. Nevertheless, SWI/SNF inhibition induced profound therapeutic response in an immunocompetent AML mouse model as well as in primary human AML samples. In vivo, SWI/SNF inhibition promoted leukemic differentiation and reduced the leukemic stem cell burden in bone marrow but also induced leukopenia. These results reveal a variable therapeutic window for SWI/SNF blockade in AML and highlight important off-tumor effects of such therapies in immunocompetent settings.
Significance: Disruption of PU.1-directed enhancer programs upon SWI/SNF inhibition causes differentiation of AML cells and induces leukopenia of PU.1-dependent B cells and monocytes, revealing the on- and off-tumor effects of SWI/SNF blockade.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007;7:105–17. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
