

TMEM161B regulates cerebral cortical gyration, Sonic Hedgehog signaling, and ciliary structure in the developing central nervous system
- PMID: 36669111
- PMCID: PMC9942790
- DOI: 10.1073/pnas.2209964120
TMEM161B regulates cerebral cortical gyration, Sonic Hedgehog signaling, and ciliary structure in the developing central nervous system
Abstract
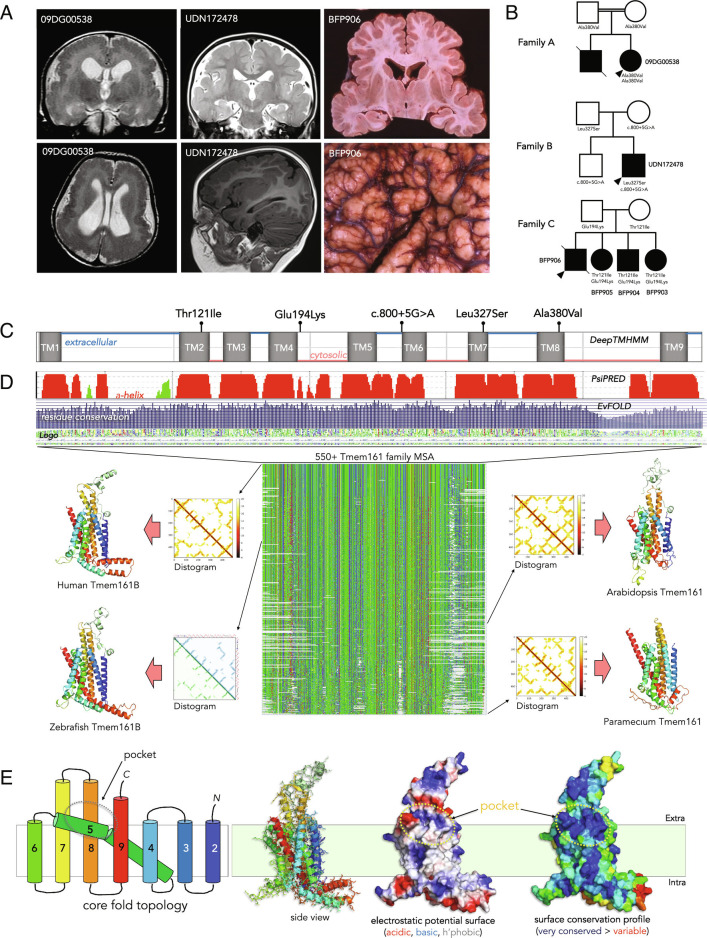
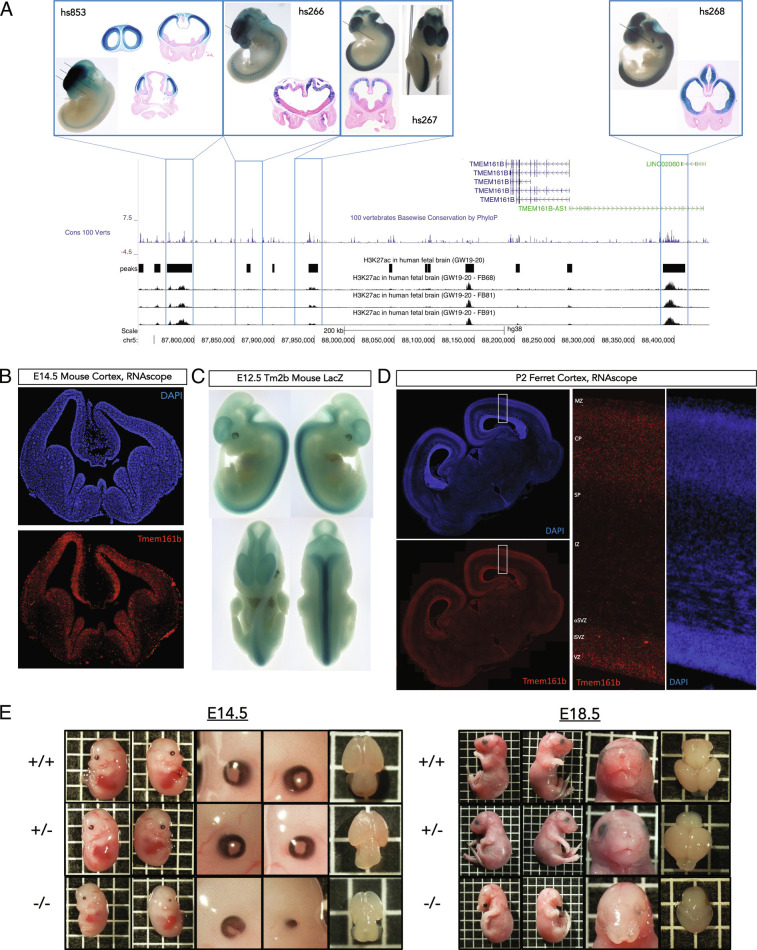
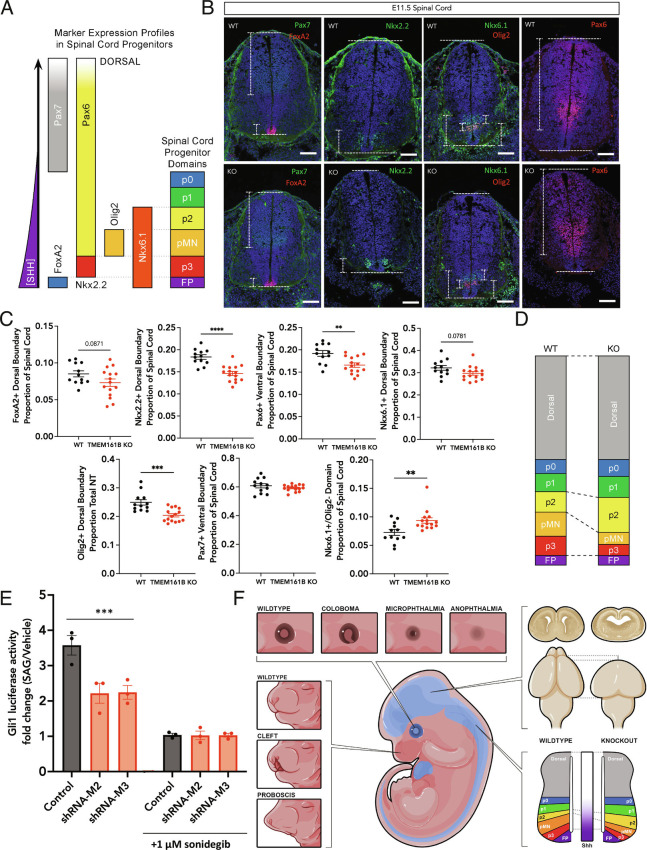
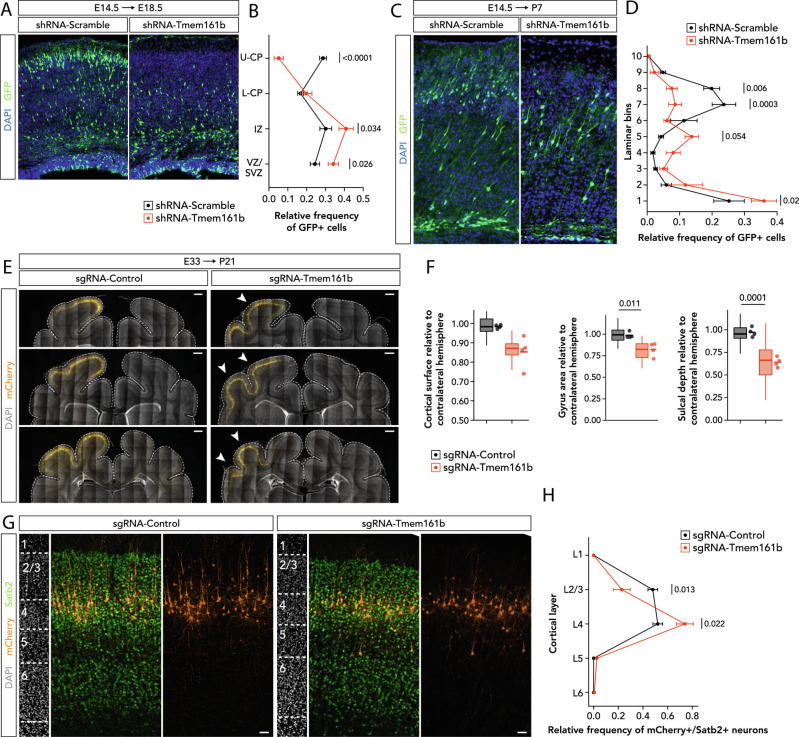
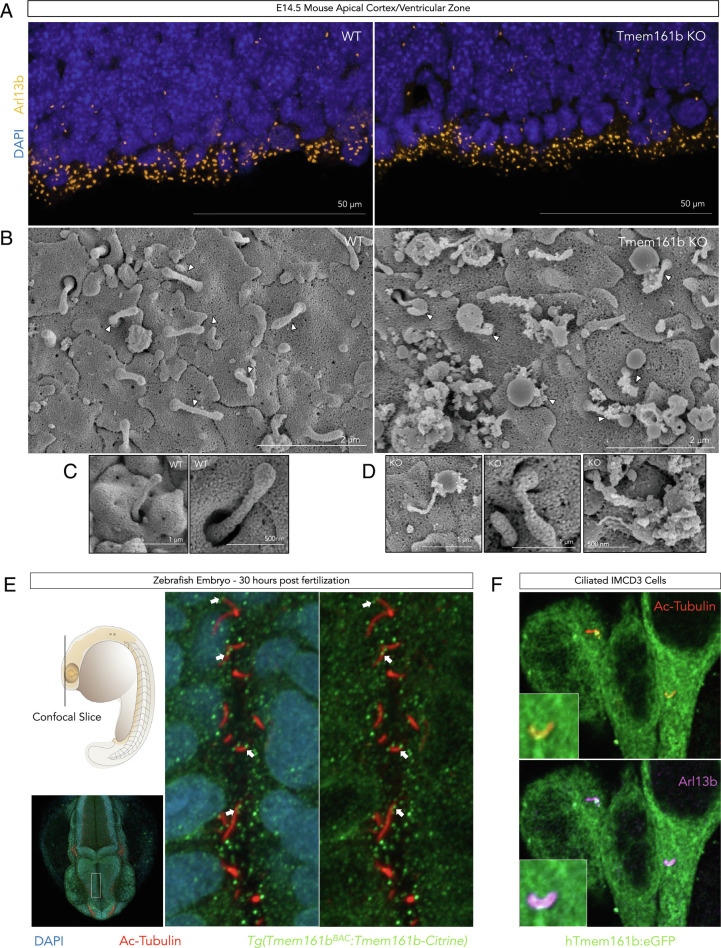
Sonic hedgehog signaling regulates processes of embryonic development across multiple tissues, yet factors regulating context-specific Shh signaling remain poorly understood. Exome sequencing of families with polymicrogyria (disordered cortical folding) revealed multiple individuals with biallelic deleterious variants in TMEM161B, which encodes a multi-pass transmembrane protein of unknown function. Tmem161b null mice demonstrated holoprosencephaly, craniofacial midline defects, eye defects, and spinal cord patterning changes consistent with impaired Shh signaling, but were without limb defects, suggesting a CNS-specific role of Tmem161b. Tmem161b depletion impaired the response to Smoothened activation in vitro and disrupted cortical histogenesis in vivo in both mouse and ferret models, including leading to abnormal gyration in the ferret model. Tmem161b localizes non-exclusively to the primary cilium, and scanning electron microscopy revealed shortened, dysmorphic, and ballooned ventricular zone cilia in the Tmem161b null mouse, suggesting that the Shh-related phenotypes may reflect ciliary dysfunction. Our data identify TMEM161B as a regulator of cerebral cortical gyration, as involved in primary ciliary structure, as a regulator of Shh signaling, and further implicate Shh signaling in human gyral development.
Keywords: Sonic Hedgehog; cortical gyration; holoprosencephaly; polymicrogyria; primary cilia.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Traiffort E., Angot E., Ruat M., Sonic Hedgehog signaling in the mammalian brain. J. Neurochem. 113 , 576–590 (2010). - PubMed
-
- Andreu-Cervera A., Catala M., Schneider-Maunoury S., Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol. Dis. 150 , 105236 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HG006348/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- T32 EB001680/EB/NIBIB NIH HHS/United States
- U01 HG007709/HG/NHGRI NIH HHS/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- P30 CA015083/CA/NCI NIH HHS/United States
- R01 NS035129/NS/NINDS NIH HHS/United States
- R01 NS100007/NS/NINDS NIH HHS/United States
- MC_UP_2201/1/MRC_/Medical Research Council/United Kingdom
- P50 HD105351/HD/NICHD NIH HHS/United States
- T32 AG000222/AG/NIA NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 MH020017/MH/NIMH NIH HHS/United States
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- MC_UP_1502/3/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
