

Benchmarking of two bioinformatic workflows for the analysis of whole-genome sequenced Staphylococcus aureus collected from patients with suspected sepsis
- PMID: 36670352
- PMCID: PMC9863170
- DOI: 10.1186/s12879-022-07977-0
Benchmarking of two bioinformatic workflows for the analysis of whole-genome sequenced Staphylococcus aureus collected from patients with suspected sepsis
Abstract
Background: The rapidly growing area of sequencing technologies, and more specifically bacterial whole-genome sequencing, could offer applications in clinical microbiology, including species identification of bacteria, prediction of genetic antibiotic susceptibility and virulence genes simultaneously. To accomplish the aforementioned points, the commercial cloud-based platform, 1928 platform (1928 Diagnostics, Gothenburg, Sweden) was benchmarked against an in-house developed bioinformatic pipeline as well as to reference methods in the clinical laboratory.
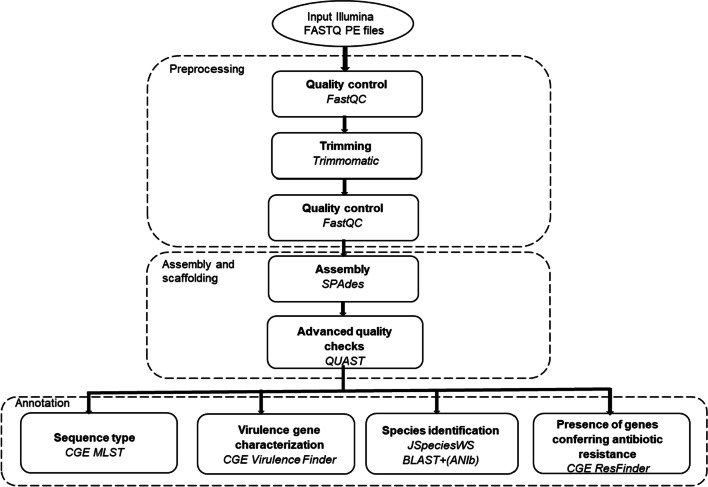
Methods: Whole-genome sequencing data retrieved from 264 Staphylococcus aureus isolates using the Illumina HiSeq X next-generation sequencing technology was used. The S. aureus isolates were collected during a prospective observational study of community-onset severe sepsis and septic shock in adults at Skaraborg Hospital, in the western region of Sweden. The collected isolates were characterized according to accredited laboratory methods i.e., species identification by MALDI-TOF MS analysis and phenotypic antibiotic susceptibility testing (AST) by following the EUCAST guidelines. Concordance between laboratory methods and bioinformatic tools, as well as concordance between the bioinformatic tools was assessed by calculating the percent of agreement.
Results: There was an overall high agreement between predicted genotypic AST and phenotypic AST results, 98.0% (989/1006, 95% CI 97.3-99.0). Nevertheless, the 1928 platform delivered predicted genotypic AST results with lower very major error rates but somewhat higher major error rates compared to the in-house pipeline. There were differences in processing times i.e., minutes versus hours, where the 1928 platform delivered the results faster. Furthermore, the bioinformatic workflows showed overall 99.4% (1267/1275, 95% CI 98.7-99.7) agreement in genetic prediction of the virulence gene characteristics and overall 97.9% (231/236, 95% CI 95.0-99.2%) agreement in predicting the sequence types (ST) of the S. aureus isolates.
Conclusions: Altogether, the benchmarking disclosed that both bioinformatic workflows are able to deliver results with high accuracy aiding diagnostics of severe infections caused by S. aureus. It also illustrates the need of international agreement on quality control and metrics to facilitate standardization of analytical approaches for whole-genome sequencing based predictions.
Keywords: Antimicrobial susceptibility; Benchmarking; Clinical microbiology; Illumina sequencing; S. aureus; Species identification; Virulence genes; Whole-genome sequencing.
© 2023. The Author(s).
Conflict of interest statement
HE was employed by the company Unilabs. DA was employed by the company 1928 Diagnostics. DT, AP, AT, MS and SJ do not have any competing interests.
Figures

References
-
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. - DOI - PMC - PubMed
-
- Garnacho-Montero J, Huici-Moreno MJ, Gutierrez-Pizarraya A, Lopez I, Marquez-Vacaro JA, Macher H, et al. Prognostic and diagnostic value of eosinopenia, C-reactive protein, procalcitonin, and circulating cell-free DNA in critically ill patients admitted with suspicion of sepsis. Crit Care. 2014;18(3):R116. doi: 10.1186/cc13908. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
