

A Customized Monkeypox Virus Genomic Database (MPXV DB v1.0) for Rapid Sequence Analysis and Phylogenomic Discoveries in CLC Microbial Genomics
- PMID: 36680080
- PMCID: PMC9861985
- DOI: 10.3390/v15010040
A Customized Monkeypox Virus Genomic Database (MPXV DB v1.0) for Rapid Sequence Analysis and Phylogenomic Discoveries in CLC Microbial Genomics
Abstract
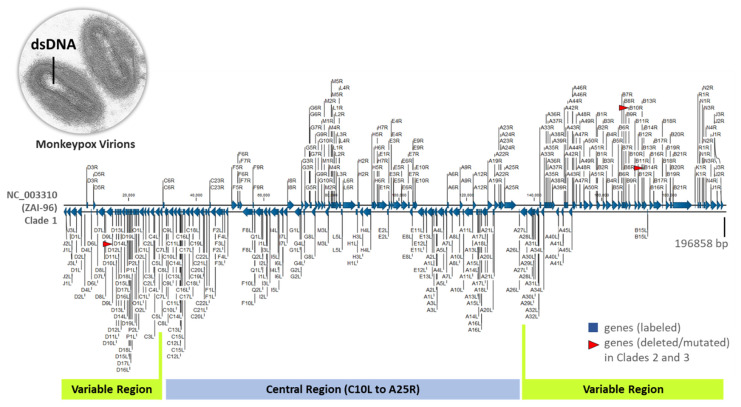
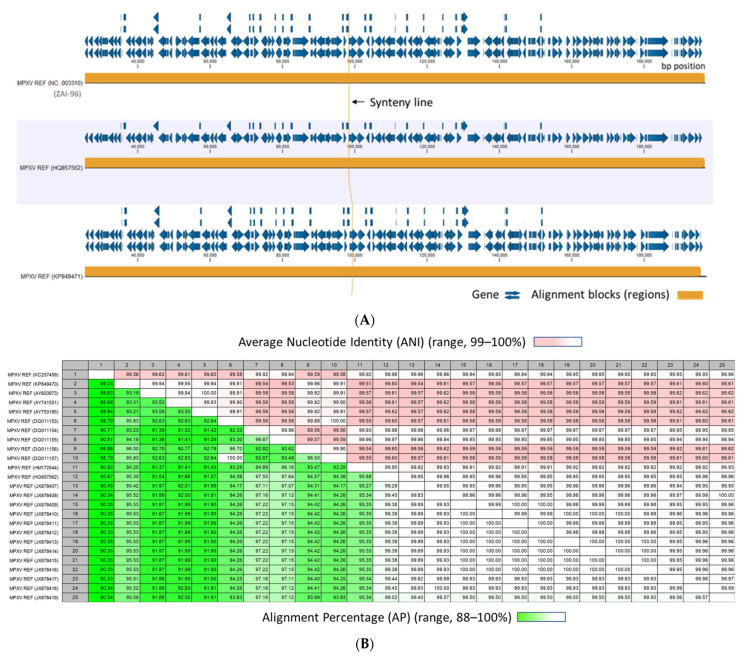
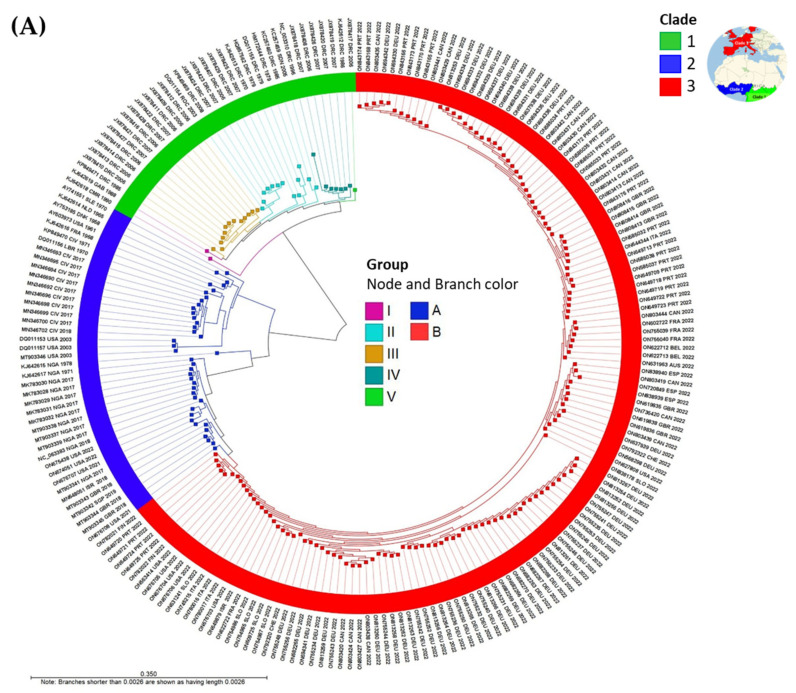
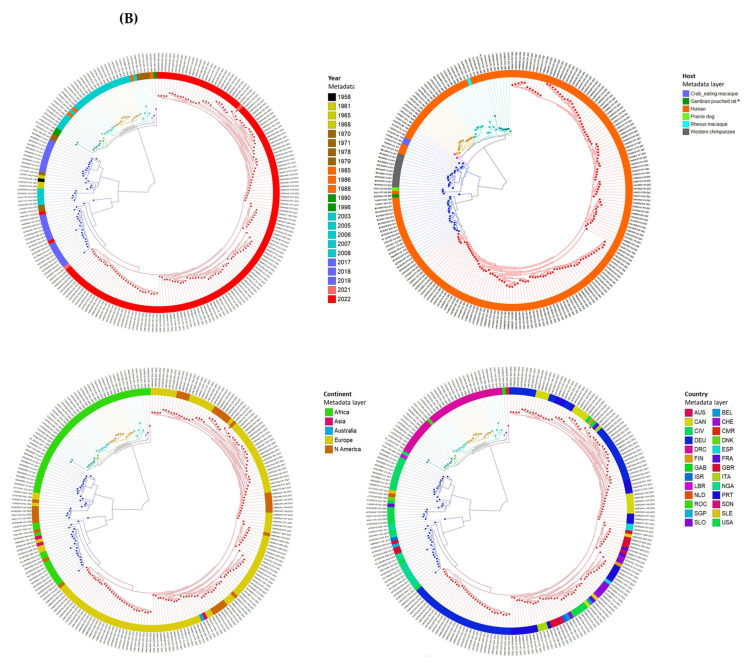
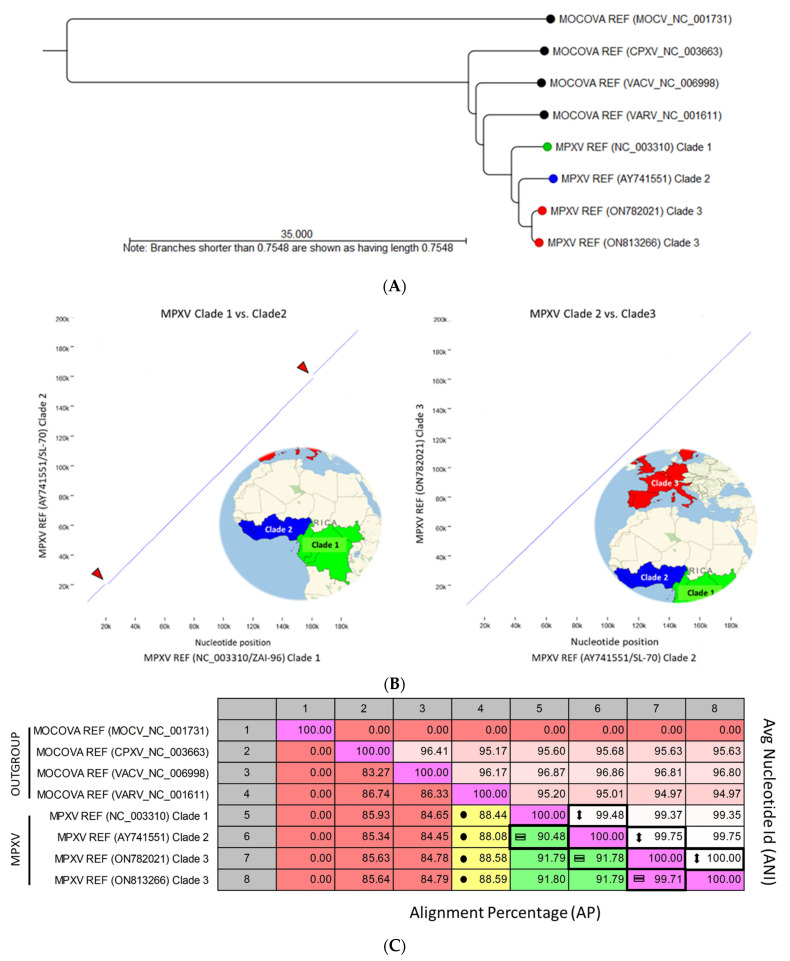
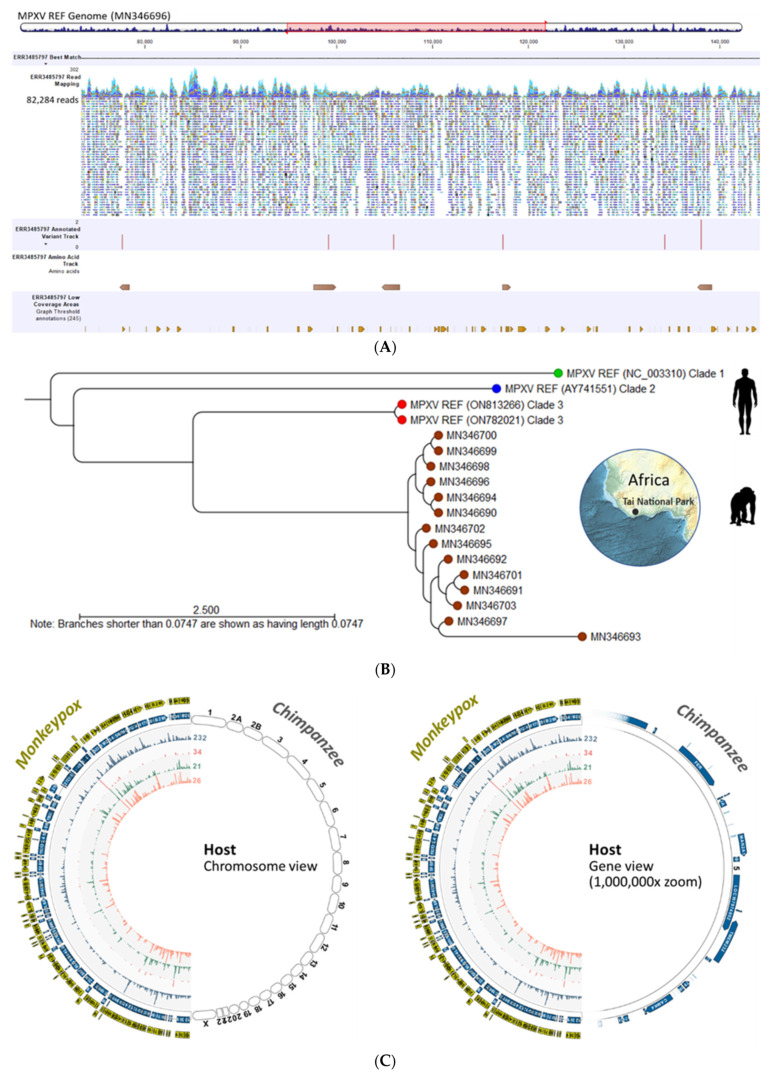
Monkeypox has been a neglected, zoonotic tropical disease for over 50 years. Since the 2022 global outbreak, hundreds of human clinical samples have been subjected to next-generation sequencing (NGS) worldwide with raw data deposited in public repositories. However, sequence analysis for in-depth investigation of viral evolution remains hindered by the lack of a curated, whole genome Monkeypox virus (MPXV) database (DB) and efficient bioinformatics pipelines. To address this, we developed a customized MPXV DB for integration with "ready-to-use" workflows in the CLC Microbial Genomics Module for whole genomic and metagenomic analysis. After database construction (218 MPXV genomes), whole genome alignment, pairwise comparison, and evolutionary analysis of all genomes were analyzed to autogenerate tabular outputs and visual displays (collective runtime: 16 min). The clinical utility of the MPXV DB was demonstrated by using a Chimpanzee fecal, hybrid-capture NGS dataset (publicly available) for metagenomic, phylogenomic, and viral/host integration analysis. The clinically relevant MPXV DB embedded in CLC workflows proved to be a rapid method of sequence analysis useful for phylogenomic exploration and a wide range of applications in translational science.
Keywords: bioinformatics; disease outbreaks; monkeypox; monkeypox virus; next generation sequencing; phylogeny; poxvirus; taxonomic classification; virus database.
Conflict of interest statement
The Defense Health Agency (DHA) of the U.S. Department of Defense has licensed the customized MPXV v1.0 and MOCVA v1.0 databases described herein to QIAGEN Digital Insights. The inventor of the customized taxonomy is J.SG. No potential conflicts of interest were disclosed by the other authors. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. This paper has undergone PAO review at Brooke Army Medical Center and was cleared for publication. The view(s) expressed herein are those of the authors and do not reflect the official policy or position of Brooke Army Medical Center, the United States Army Medical Department, the United States Army Office of the Surgeon General, the Department of the Army, the Defense Health Agency, the Department of Defense or the United States Government.
Figures

Similar articles
-
Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology.Int J Mol Sci. 2025 Feb 8;26(4):1428. doi: 10.3390/ijms26041428. Int J Mol Sci. 2025. PMID: 40003895 Free PMC article.
-
A comprehensive review of monkeypox virus and mpox characteristics.Front Cell Infect Microbiol. 2024 Mar 6;14:1360586. doi: 10.3389/fcimb.2024.1360586. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38510963 Free PMC article. Review.
-
Monkeypox virus (MPXV) genomics: A mutational and phylogenomic analyses of B.1 lineages.Travel Med Infect Dis. 2023 Mar-Apr;52:102551. doi: 10.1016/j.tmaid.2023.102551. Epub 2023 Feb 23. Travel Med Infect Dis. 2023. PMID: 36746267 Free PMC article.
-
Exploring the key genomic variation in monkeypox virus during the 2022 outbreak.BMC Genom Data. 2023 Nov 16;24(1):67. doi: 10.1186/s12863-023-01171-0. BMC Genom Data. 2023. PMID: 37968621 Free PMC article.
-
Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution.Viruses. 2020 Nov 5;12(11):1257. doi: 10.3390/v12111257. Viruses. 2020. PMID: 33167496 Free PMC article. Review.
Cited by
-
Genomic and Antigenic Differences Between Monkeypox Virus and Vaccinia Vaccines: Insights and Implications for Vaccinology.Int J Mol Sci. 2025 Feb 8;26(4):1428. doi: 10.3390/ijms26041428. Int J Mol Sci. 2025. PMID: 40003895 Free PMC article.
-
Correlation between the gut microbiome and neurodegenerative diseases: a review of metagenomics evidence.Neural Regen Res. 2024 Apr;19(4):833-845. doi: 10.4103/1673-5374.382223. Neural Regen Res. 2024. PMID: 37843219 Free PMC article. Review.
-
Molecular Diagnosis of Human Monkeypox Virus during 2022-23 Outbreak: Preliminary Evaluation of Novel Real-Time Qualitative PCR Assays.Microorganisms. 2024 Mar 27;12(4):664. doi: 10.3390/microorganisms12040664. Microorganisms. 2024. PMID: 38674608 Free PMC article.
-
A comprehensive review of monkeypox virus and mpox characteristics.Front Cell Infect Microbiol. 2024 Mar 6;14:1360586. doi: 10.3389/fcimb.2024.1360586. eCollection 2024. Front Cell Infect Microbiol. 2024. PMID: 38510963 Free PMC article. Review.
-
Nosocomial transmission of fluconazole-resistant Candida glabrata bloodstream isolates revealed by whole-genome sequencing.Microbiol Spectr. 2024 Oct 3;12(10):e0088324. doi: 10.1128/spectrum.00883-24. Epub 2024 Aug 20. Microbiol Spectr. 2024. PMID: 39162519 Free PMC article.
References
-
- Magnus P.V., Andersen E.K., Petersen K.B., Birch-Andersen A. A pox-like disease in cynomolgus monkeys. Acta Pathol. Microbiol. Scand. 1959;46:156–176. doi: 10.1111/j.1699-0463.1959.tb00328.x. - DOI
-
- World Health Organization (WHO) [(accessed on 10 November 2022)]. Available online: https://www.who.int/director-general/speeches/detail/who-director-genera....
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
