

The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles
- PMID: 36680147
- PMCID: PMC9861436
- DOI: 10.3390/v15010107
The Effect of Treatment-Associated Mutations on HIV Replication and Transmission Cycles
Abstract
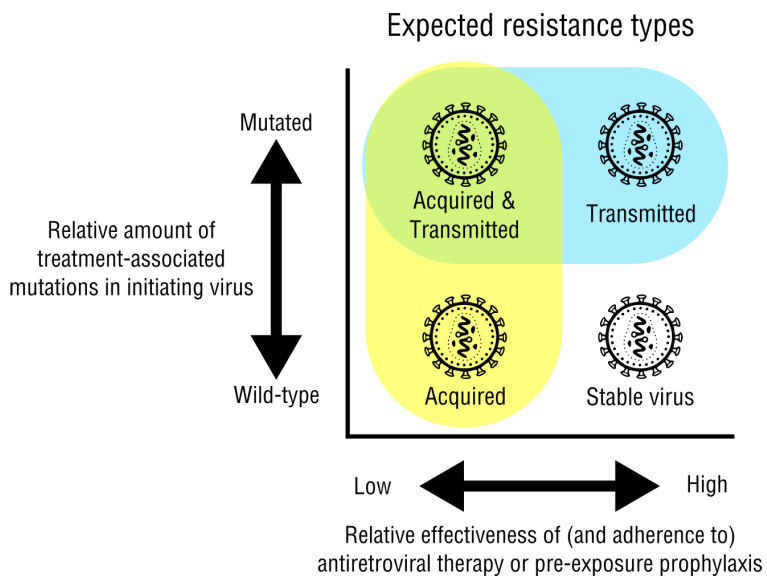
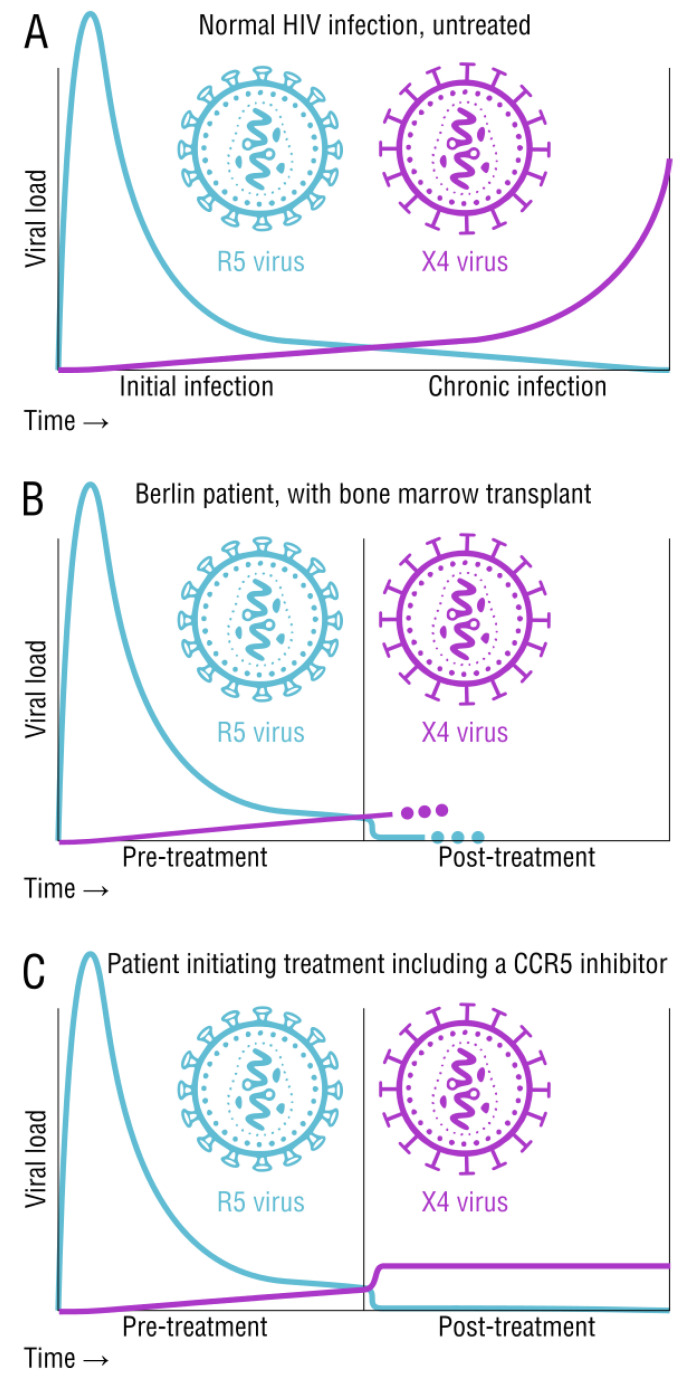
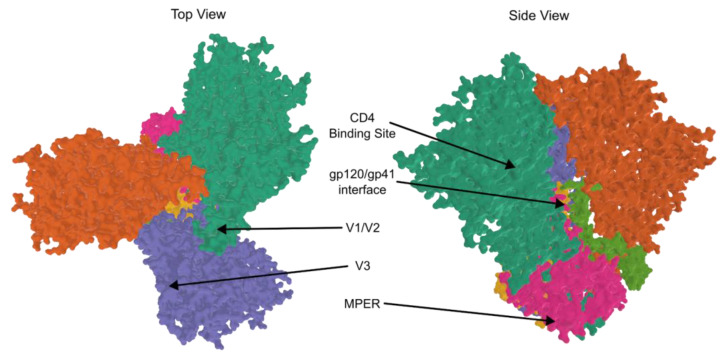
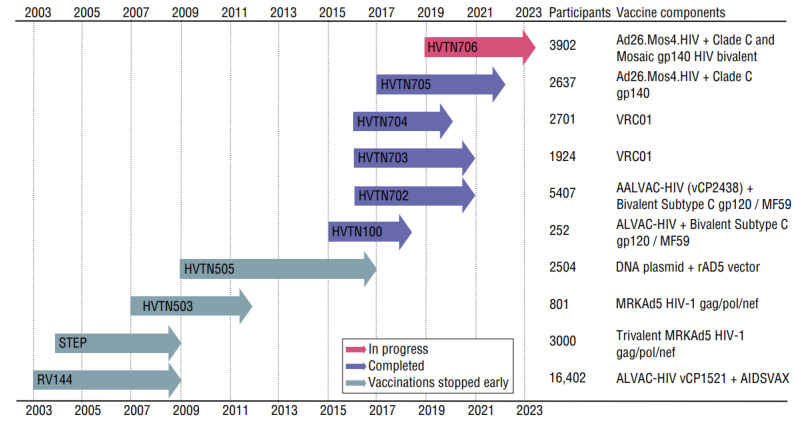
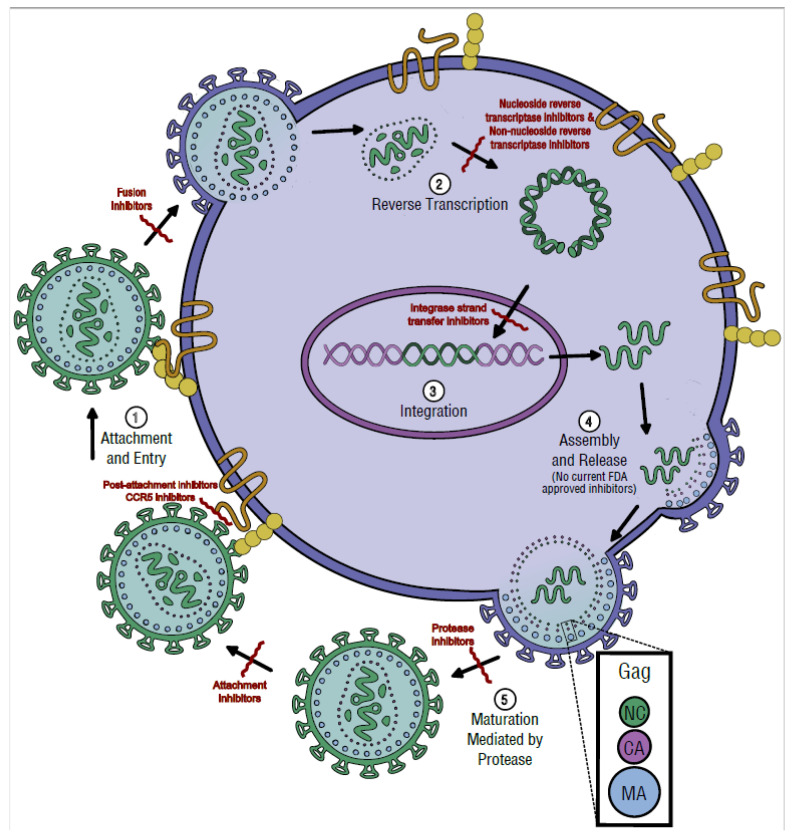
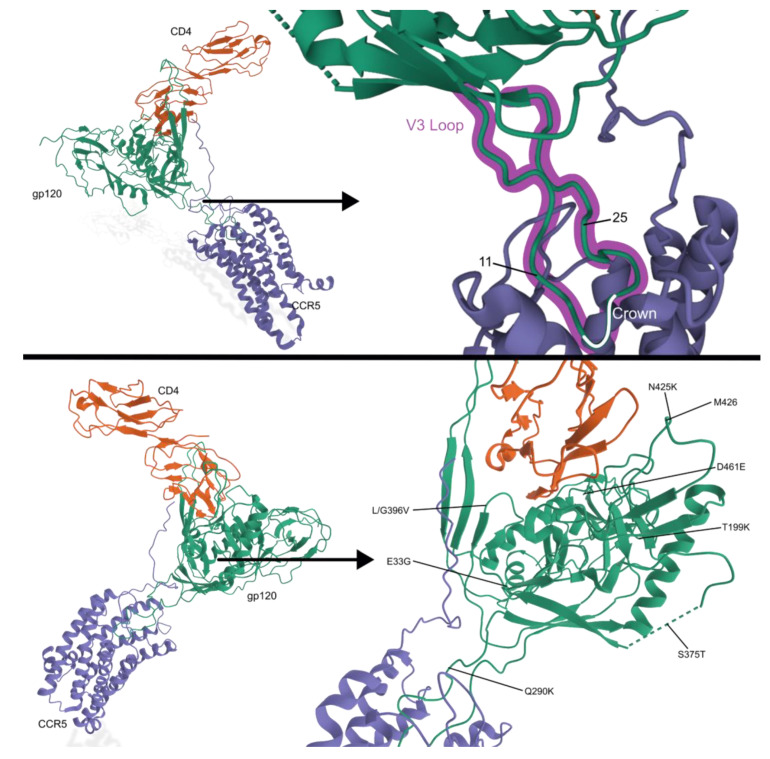
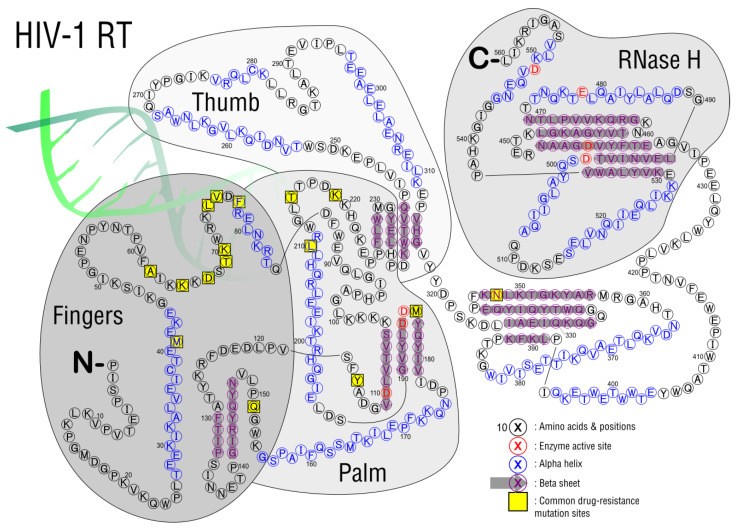
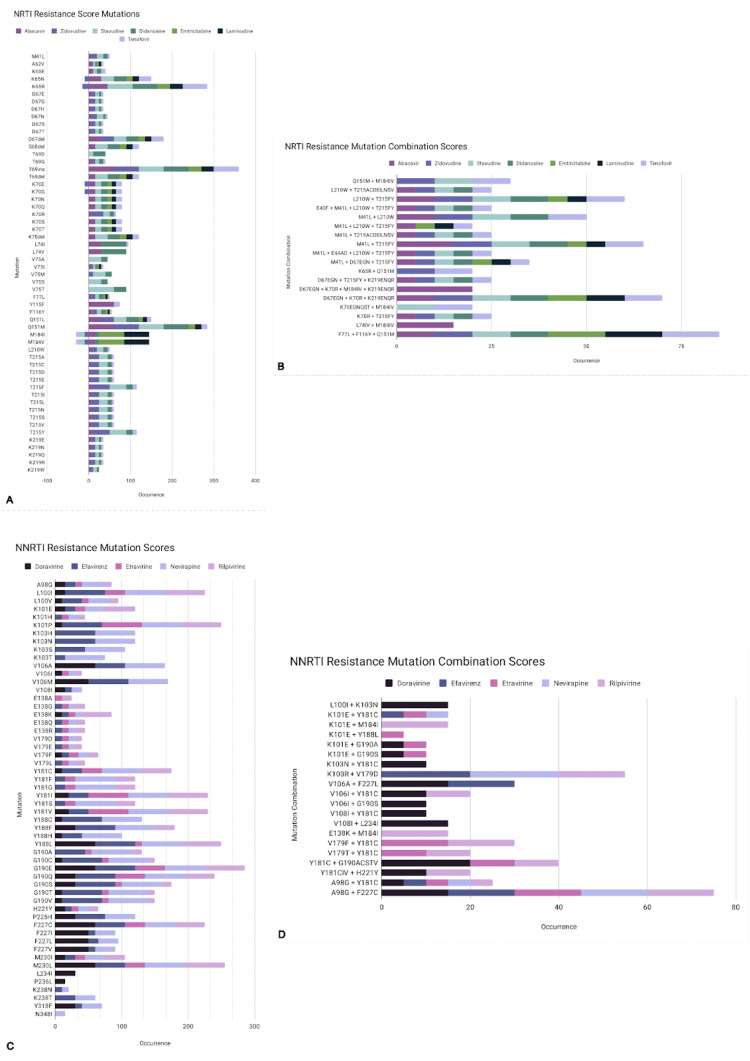
HIV/AIDS mortality has been decreasing over the last decade. While promising, this decrease correlated directly with increased use of antiretroviral drugs. As a natural consequence of its high mutation rate, treatments provide selection pressure that promotes the natural selection of escape mutants. Individuals may acquire drug-naive strains, or those that have already mutated due to treatment. Even within a host, mutation affects HIV tropism, where initial infection begins with R5-tropic virus, but the clinical transition to AIDS correlates with mutations that lead to an X4-tropic switch. Furthermore, the high mutation rate of HIV has spelled failure for all attempts at an effective vaccine. Pre-exposure drugs are currently the most effective drug-based preventatives, but their effectiveness is also threatened by viral mutation. From attachment and entry to assembly and release, the steps in the replication cycle are also discussed to describe the drug mechanisms and mutations that arise due to those drugs. Revealing the patterns of HIV-1 mutations, their effects, and the coordinated attempt to understand and control them will lead to effective use of current preventative measures and treatment options, as well as the development of new ones.
Keywords: HIV-1; drug resistance; mutation; vaccines.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Study of the impact of HIV genotypic drug resistance testing on therapy efficacy.Verh K Acad Geneeskd Belg. 2001;63(5):447-73. Verh K Acad Geneeskd Belg. 2001. PMID: 11813503 Review.
-
Deep-Sequencing Analysis of the Dynamics of HIV-1 Quasiespecies in Naive Patients during a Short Exposure to Maraviroc.J Virol. 2018 May 14;92(11):e00390-18. doi: 10.1128/JVI.00390-18. Print 2018 Jun 1. J Virol. 2018. PMID: 29563289 Free PMC article. Clinical Trial.
-
HIV-1 dual/mixed tropic isolates show different genetic and phenotypic characteristics and response to maraviroc in vitro.Antiviral Res. 2011 Apr;90(1):42-53. doi: 10.1016/j.antiviral.2011.02.005. Epub 2011 Feb 22. Antiviral Res. 2011. PMID: 21349294
-
Inhibition of dual/mixed tropic HIV-1 isolates by CCR5-inhibitors in primary lymphocytes and macrophages.PLoS One. 2013 Jul 9;8(7):e68076. doi: 10.1371/journal.pone.0068076. Print 2013. PLoS One. 2013. PMID: 23874501 Free PMC article.
-
The challenge of antiretroviral-drug-resistant HIV: is there any possible clinical advantage?Curr HIV Res. 2004 Jul;2(3):283-92. doi: 10.2174/1570162043351192. Curr HIV Res. 2004. PMID: 15279592 Review.
Cited by
-
Treatment Management Challenges in Naïve and Experienced HIV-1-Infected Individuals Carrying the M184V Mutation.Viruses. 2024 Aug 30;16(9):1392. doi: 10.3390/v16091392. Viruses. 2024. PMID: 39339868 Free PMC article. Review.
-
Advances in HIV Treatment and Vaccine Development: Emerging Therapies and Breakthrough Strategies for Long-Term Control.AIDS Res Treat. 2025 Jul 4;2025:6829446. doi: 10.1155/arat/6829446. eCollection 2025. AIDS Res Treat. 2025. PMID: 40655875 Free PMC article. Review.
-
Involvement of a Rarely Used Splicing SD2b Site in the Regulation of HIV-1 vif mRNA Production as Revealed by a Growth-Adaptive Mutation.Viruses. 2023 Dec 14;15(12):2424. doi: 10.3390/v15122424. Viruses. 2023. PMID: 38140666 Free PMC article.
-
Navigating the Antiretroviral Therapy Switch Conundrum: Unveiling the Dilemma of Drug Resistance and Disease Progression in HIV/AIDS.Cureus. 2024 Mar 19;16(3):e56441. doi: 10.7759/cureus.56441. eCollection 2024 Mar. Cureus. 2024. PMID: 38638795 Free PMC article. Review.
-
An integrated mutation-based immunoinformatic approach incorporating variability in epitopes: a study based on HIV subtype C.Front Immunol. 2025 May 20;16:1540253. doi: 10.3389/fimmu.2025.1540253. eCollection 2025. Front Immunol. 2025. PMID: 40463364 Free PMC article.
References
-
- Rhee S.-Y., Kantor R., Katzenstein D.A., Camacho R., Morris L., Sirivichayakul S., Jorgensen L., Brigido L.F., Schapiro J.M., Shafer R.W., et al. HIV-1 Pol Mutation Frequency by Subtype and Treatment Experience: Extension of the HIVseq Program to Seven Non-B Subtypes. AIDS. 2006;20:643–651. doi: 10.1097/01.aids.0000216363.36786.2b. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
