

Comparative genome analysis reveals high-level drug resistance markers in a clinical isolate of Mycobacterium fortuitum subsp . fortuitum MF GZ001
- PMID: 36683685
- PMCID: PMC9846761
- DOI: 10.3389/fcimb.2022.1056007
Comparative genome analysis reveals high-level drug resistance markers in a clinical isolate of Mycobacterium fortuitum subsp . fortuitum MF GZ001
Abstract
Introduction: Infections caused by non-tuberculosis mycobacteria are significantly worsening across the globe. M. fortuitum complex is a rapidly growing pathogenic species that is of clinical relevance to both humans and animals. This pathogen has the potential to create adverse effects on human healthcare.
Methods: The MF GZ001 clinical strain was collected from the sputum of a 45-year-old male patient with a pulmonary infection. The morphological studies, comparative genomic analysis, and drug resistance profiles along with variants detection were performed in this study. In addition, comparative analysis of virulence genes led us to understand the pathogenicity of this organism.
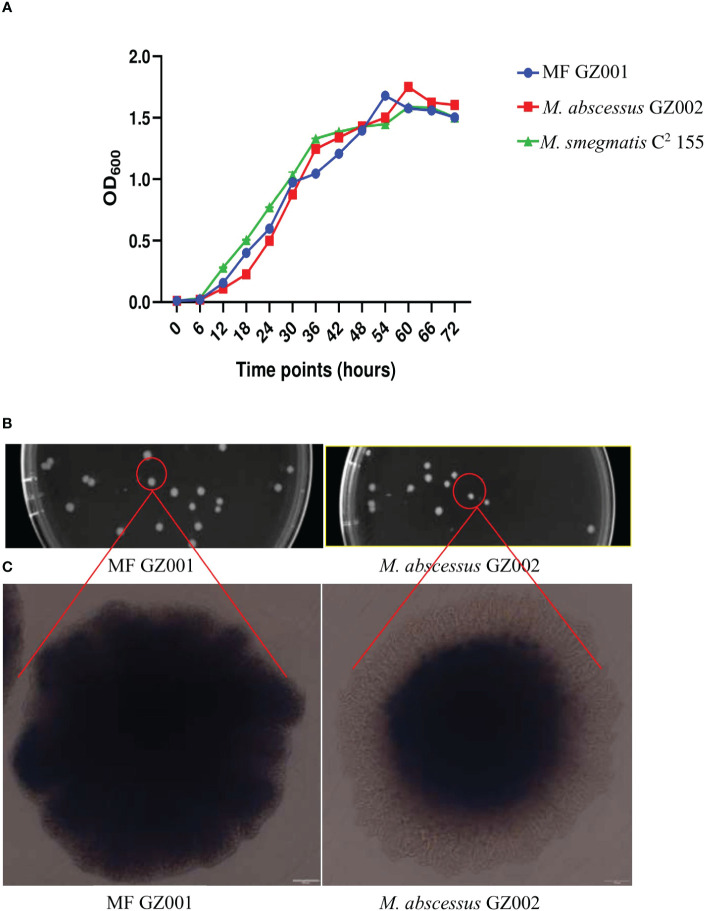
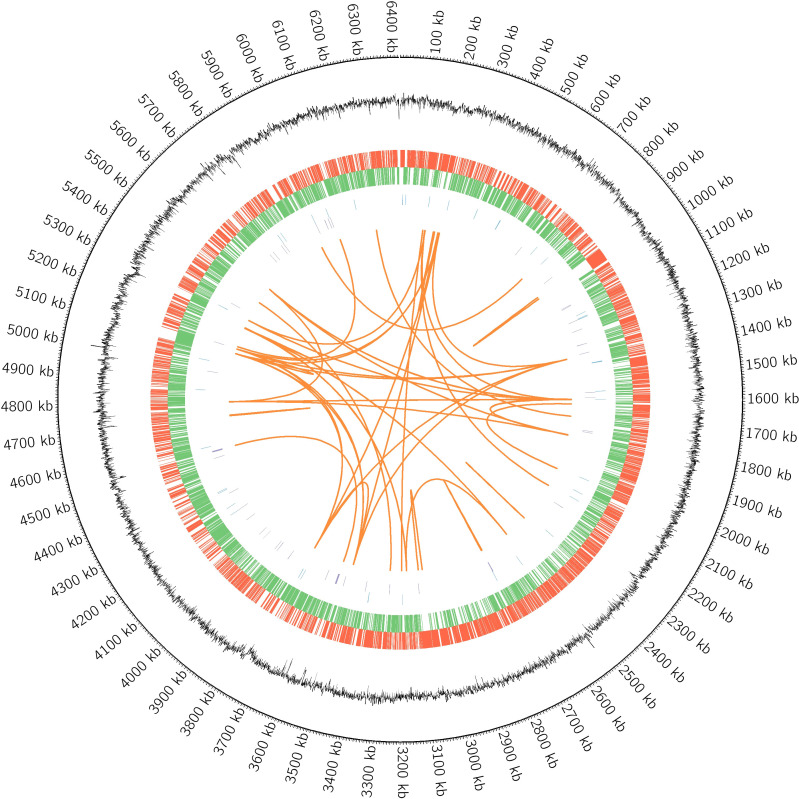
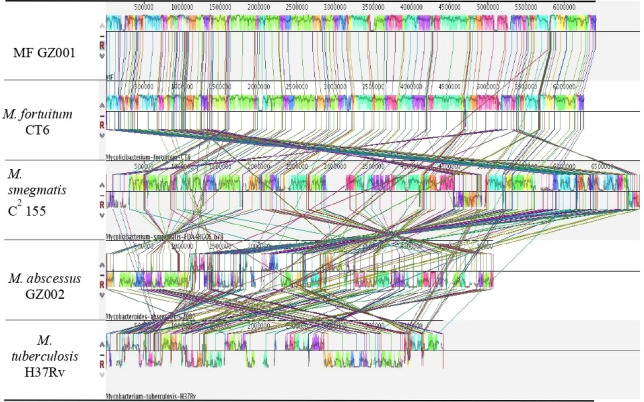
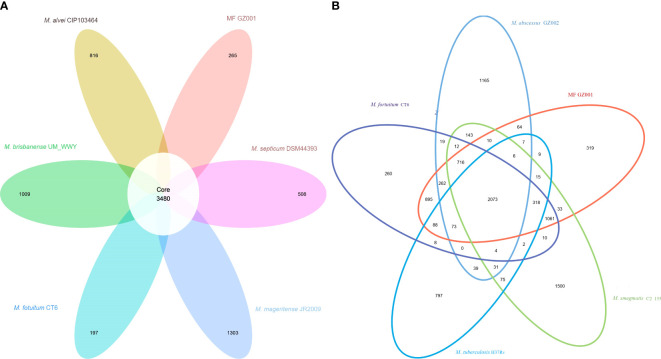
Results: Bacterial growth kinetics and morphology confirmed that MF GZ001 is a rapidly growing species with a rough morphotype. The MF GZ001 contains 6413573 bp genome size with 66.18 % high G+C content. MF GZ001 possesses a larger genome than other related mycobacteria and included 6156 protein-coding genes. Molecular phylogenetic tree, collinearity, and comparative genomic analysis suggested that MF GZ001 is a novel member of the M. fortuitum complex. We carried out the drug resistance profile analysis and found single nucleotide polymorphism (SNP) mutations in key drug resistance genes such as rpoB, katG, AAC(2')-Ib, gyrA, gyrB, embB, pncA, blaF, thyA, embC, embR, and iniA. In addition, the MF GZ001strain contains mutations in iniA, iniC, pncA, and ribD which conferred resistance to isoniazid, ethambutol, pyrazinamide, and para-aminosalicylic acid respectively, which are not frequently observed in rapidly growing mycobacteria. A wide variety of predicted putative potential virulence genes were found in MF GZ001, most of which are shared with well-recognized mycobacterial species with high pathogenic profiles such as M. tuberculosis and M. abscessus.
Discussion: Our identified novel features of a pathogenic member of the M. fortuitum complex will provide the foundation for further investigation of mycobacterial pathogenicity and effective treatment.
Keywords: Mycobacterium fortuitum; comparative genomic analysis; drug resistance; morphology; pathogenesis.
Copyright © 2023 Alam, Guan, Zhu, Zeng, Fang, Wang, Yusuf, Zhang, Tian, Fang, Gao, Khatun, Liu, Hameed, Tan, Hu, Liu and Zhang.
Conflict of interest statement
The authors declare that the research was carried out in the absence of any financial or economic relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Minor contribution of mutations at iniA codon 501 and embC-embA intergenic region in ethambutol-resistant clinical Mycobacterium tuberculosis isolates in Kuwait.Ann Clin Microbiol Antimicrob. 2009 Jan 15;8:2. doi: 10.1186/1476-0711-8-2. Ann Clin Microbiol Antimicrob. 2009. PMID: 19146672 Free PMC article.
-
katGI and katGII encode two different catalases-peroxidases in Mycobacterium fortuitum.J Bacteriol. 1997 Nov;179(22):6880-6. doi: 10.1128/jb.179.22.6880-6886.1997. J Bacteriol. 1997. PMID: 9371430 Free PMC article.
-
Antimicrobial susceptibility testing of rapidly growing mycobacteria isolated in Japan.BMC Infect Dis. 2017 Mar 7;17(1):197. doi: 10.1186/s12879-017-2298-8. BMC Infect Dis. 2017. PMID: 28270102 Free PMC article.
-
[Molecular identification of mycobacteria and detection of antibiotic resistance].Ann Biol Clin (Paris). 2004 Jul-Aug;62(4):405-13. Ann Biol Clin (Paris). 2004. PMID: 15297234 Review. French.
-
General characteristics, features of cultivation and antibiotic resistance representatives of mycobacterium fortuitum group representatives (review of literature).Klin Lab Diagn. 2021 Apr 17;66(4):223-228. doi: 10.51620/0869-2084-2021-66-4-223-228. Klin Lab Diagn. 2021. PMID: 33878244 Review. English.
Cited by
-
The acetylation of pknH is linked to the ethambutol resistance of Mycobacterium tuberculosis.Arch Microbiol. 2023 Sep 23;205(10):337. doi: 10.1007/s00203-023-03676-9. Arch Microbiol. 2023. PMID: 37740776
References
-
- Aitken M. L., Limaye A., Pottinger P., Whimbey E., Goss C. H., Tonelli M. R., et al. (2012). Respiratory outbreak of Mycobacterium abscessus subspecies massiliense in a lung transplant and cystic fibrosis center. Am. J. Respir. Crit. Care Med. 185 (2), 231–232. doi: 10.1164/ajrccm.185.2.231 - DOI - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical