

Cohort of Phenotype, Genotype, and Outcome of SCN Developmental and Epileptic Encephalopathies from Southern Part of India
- PMID: 36684540
- PMCID: PMC9848768
- DOI: 10.1055/s-0041-1731020
Cohort of Phenotype, Genotype, and Outcome of SCN Developmental and Epileptic Encephalopathies from Southern Part of India
Abstract
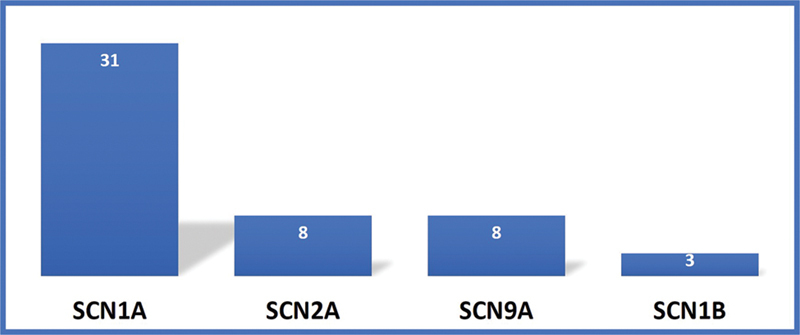
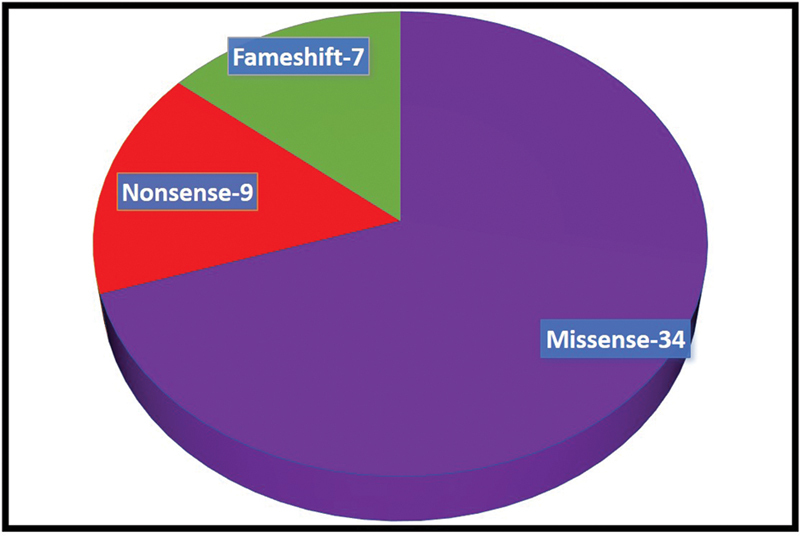
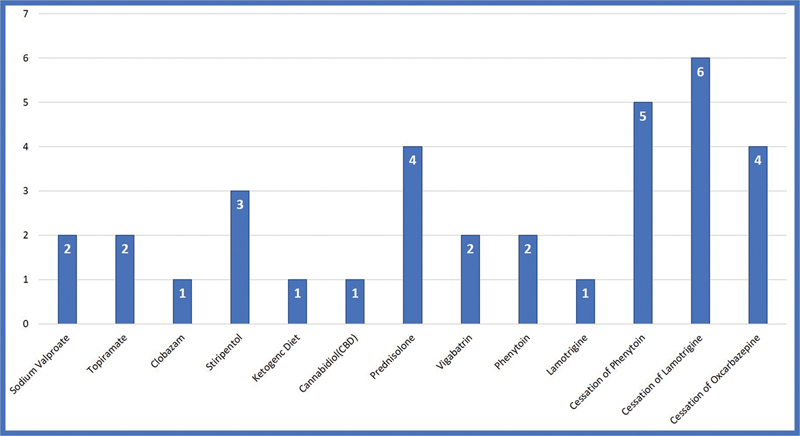
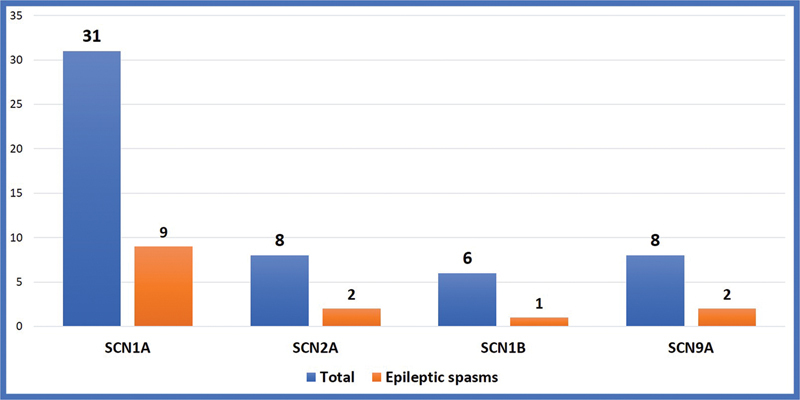
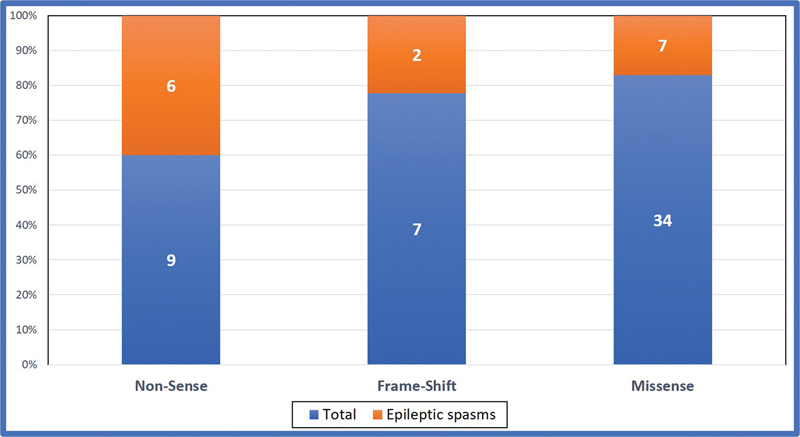
The SCN encephalopathies are one of the rare early childhood intractable epileptic encephalopathies associated with pleomorphic seizures, cognitive decline, motor, and behavioral abnormalities that begin in early infancy. There is a dearth of data on phenotype and genotype of SCN encephalopathies from the Indian subcontinent, hence we are reporting clinical and molecular profile and outcome of SCN developmental and epileptic encephalopathies. This is a retrospective chart review of SCN developmental and epileptic encephalopathies in a tertiary care center, Bangalore, India between January 2015 and March 2020. All children with clinical features of SCN developmental and epileptic encephalopathies and confirmed with pathogenic variants were included. A total of 50 cases of SCN developmental and epileptic encephalopathies were analyzed, 31 of them were male and the mean age of presentation was 7.8 months. Precipitating factors for the first episode of seizure were fever and vaccination accounting for 33 and 8 children, respectively. Forty (80%) children had prolonged seizures and 15 (30%) had epileptic spasms. All children had a normal birth history and normal development before the onset of seizures, which was followed by developmental delay and regression. Thirty (60%) children had behavioral difficulties, notable hyperactivity, and autistic features. Neuroimaging and the initial electroencephalogram (EEG) were normal in all patients. The mean age of abnormal EEG was 14 months. The various subtypes of SCN variants were SCN1A in 31 children followed by SCN2A and SCN9A in eight children each and SCN1B in three children. Frameshift and nonsense mutations were associated with more severe phenotype and poor outcome compared with missense mutations. Thirty-four patients partially responded to treatment and the rest were refractory. The results of genetic testing were used to guide treatment; sodium channel blocking antiepileptic drugs were discontinued in 15 patients and sodium channel blocking agents were started in 3 patients with partial response. Three out of four children on stiripentol had a partial response. The SCN developmental and epileptic encephalopathies can present with epileptic spasms in addition to other types of seizures. Epileptic spasms are more common in nonsense and frameshift mutations. The outcome is poor in children with epileptic spasms compared with those without epileptic spasms. Genetic testing helps to select antiepileptic drugs that lead to seizure reduction.
Keywords: Dravet syndrome; SCN developmental and epileptic encephalopathies; SCN mutation; epileptic spasms.
Thieme. All rights reserved.
Conflict of interest statement
Conflict of Interest None declared.
Figures
Similar articles
-
Case Series of Early SCN1A-Related Developmental and Epileptic Encephalopathies.J Pediatr Neurosci. 2021 Jul-Sep;16(3):212-217. doi: 10.4103/jpn.JPN_99_20. Epub 2021 Jul 2. J Pediatr Neurosci. 2021. PMID: 36160609 Free PMC article.
-
Voltage-gated sodium channel epilepsies in a tertiary care center: Phenotypic spectrum with correlation to predicted functional effects.Epilepsy Behav. 2024 Sep;158:109930. doi: 10.1016/j.yebeh.2024.109930. Epub 2024 Jul 3. Epilepsy Behav. 2024. PMID: 38964184
-
A retrospective study of the yield of next-generation sequencing in the diagnosis of developmental and epileptic encephalopathies and epileptic encephalopathies in 0-12 years aged children at a single tertiary care hospital in South India.Epileptic Disord. 2024 Oct;26(5):609-625. doi: 10.1002/epd2.20254. Epub 2024 Jun 24. Epileptic Disord. 2024. PMID: 38923778
-
Summary of recommendations for the management of infantile seizures: Task Force Report for the ILAE Commission of Pediatrics.Epilepsia. 2015 Aug;56(8):1185-97. doi: 10.1111/epi.13057. Epub 2015 Jun 30. Epilepsia. 2015. PMID: 26122601 Review.
-
Diagnostic Approach to Genetic Causes of Early-Onset Epileptic Encephalopathy.J Child Neurol. 2016 Mar;31(4):523-32. doi: 10.1177/0883073815599262. Epub 2015 Aug 13. J Child Neurol. 2016. PMID: 26271793 Review.
Cited by
-
Identification of five novel SCN1A variants.Front Behav Neurosci. 2023 Nov 8;17:1272748. doi: 10.3389/fnbeh.2023.1272748. eCollection 2023. Front Behav Neurosci. 2023. PMID: 38025388 Free PMC article.
References
-
- Scheffer I E, Nabbout R. SCN1A -related phenotypes: Epilepsy and beyond . Epilepsia. 2019;60 03:S17–S24. - PubMed
-
- Hattori J, Ouchida M, Ono J. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia. 2008;49(04):626–633. - PubMed
-
- Fujiwara T, Sugawara T, Mazaki-Miyazaki E. Mutations of sodium channel alpha subunit type 1 ( SCN1A ) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures Brain 2003126(Pt 3):531–546. - PubMed
-
- Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infancy: Dravet syndrome. Adv Neurol. 2005;95:71–102. - PubMed
-
- Brunklaus A, Ellis R, Reavey E, Forbes G H, Zuberi S M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome Brain 2012135(Pt 8):2329–2336. - PubMed
LinkOut - more resources
Full Text Sources
