

Case report: Pneumocystis jirovecii pneumonia in a severe case of Aicardi-Goutières syndrome with an IFIH1 gain-of-function mutation mimicking combined immunodeficiency
- PMID: 36685504
- PMCID: PMC9846174
- DOI: 10.3389/fimmu.2022.1033513
Case report: Pneumocystis jirovecii pneumonia in a severe case of Aicardi-Goutières syndrome with an IFIH1 gain-of-function mutation mimicking combined immunodeficiency
Abstract
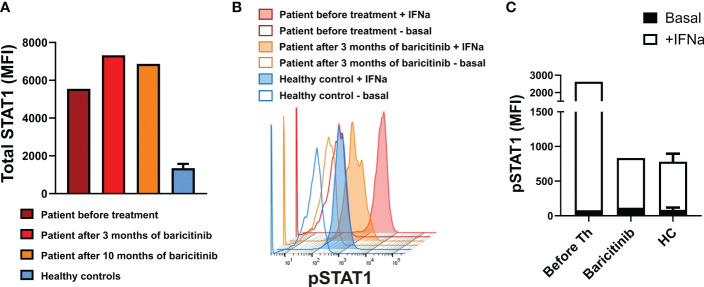
Aicardi-Goutières syndrome (AGS) is a genetically determined early-onset progressive encephalopathy caused by mutations leading to overexpression of type I interferon (IFN) and resulting in various clinical phenotypes. A gain-of-function (GOF) mutation in the IFIH1 gene is associated with robust production of type I IFN and activation of the Janus kinase (JAK) signal transducer and activator of the transcription (STAT) pathway, which can cause AGS type 7. We detail the clinical case of an infant who initially presented with Pneumocystis jirovecii pneumonia (PCP), had recurrent respiratory infections, and was later treated with a JAK inhibitor, baricitinib, because of a genetically confirmed GOF mutation in the IFIH1 gene. This spectrum of IFIH1 GOF mutations with overlapping features of hyperinflammation and severe opportunistic infection, which mimics combined immunodeficiency (CID), has not been described before. In this case, therapy with baricitinib effectively blocked IFN-α activation and reduced STAT1 signaling but had no effect on the progression of the neurological disease.
Keywords: Aicardi–Goutières syndrome (AGS); IFIH1 gene; Janus kinase inhibitor; combined immune deficiency; interferonopathy.
Copyright © 2023 Železnik, Soltirovska Šalamon, Debeljak, Goropevšek, Šuštar, Ključevšek, Ihan and Avčin.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Short-term efficacy of tofacitinib, a JAK inhibitor, in IFIH1-related Aicardi-Goutières syndrome.Eur J Med Genet. 2025 Jun;75:105006. doi: 10.1016/j.ejmg.2025.105006. Epub 2025 Mar 3. Eur J Med Genet. 2025. PMID: 40043752
-
Neurological Findings and a Brief Review of the Current Literature in a Severe Case of Aicardi-Goutières Syndrome Due to an IFIH1 Mutation.Neuropediatrics. 2024 Oct;55(5):337-340. doi: 10.1055/a-2321-0597. Epub 2024 May 7. Neuropediatrics. 2024. PMID: 38714209 Review.
-
Severe diarrhea in a 10-year-old girl with Aicardi-Goutières syndrome due to IFIH1 gene mutation.Am J Med Genet A. 2021 Oct;185(10):3146-3152. doi: 10.1002/ajmg.a.62397. Epub 2021 Jun 29. Am J Med Genet A. 2021. PMID: 34189822
-
Overlapping Aicardi-Goutières and Singleton-Merten syndromes with a heterozygous gain-of-function mutation in IFIH1 mimicking juvenile idiopathic arthritis.Immunol Med. 2025 Sep;48(3):256-260. doi: 10.1080/25785826.2025.2479148. Epub 2025 Mar 21. Immunol Med. 2025. PMID: 40116369
-
MDA5-Associated Neuroinflammation and the Singleton-Merten Syndrome: Two Faces of the Same Type I Interferonopathy Spectrum.J Interferon Cytokine Res. 2017 May;37(5):214-219. doi: 10.1089/jir.2017.0004. J Interferon Cytokine Res. 2017. PMID: 28475458 Free PMC article. Review.
Cited by
-
Experimental and clinical tests of FDA-approved kinase inhibitors for the treatment of neurological disorders (update 2024).Explor Drug Sci. 2025;3:1008116. doi: 10.37349/eds.2025.1008116. Epub 2025 Jul 1. Explor Drug Sci. 2025. PMID: 40708570 Free PMC article.
-
Baricitinib Treatment in RNU7-1-Associated Aicardi-Goutières Syndrome in a South African Child: A Case Report.Am J Med Genet A. 2025 May;197(5):e63978. doi: 10.1002/ajmg.a.63978. Epub 2025 Jan 2. Am J Med Genet A. 2025. PMID: 39748568 Free PMC article.
-
Trends in the Epidemiology of Pneumocystis Pneumonia in Immunocompromised Patients without HIV Infection.J Fungi (Basel). 2023 Jul 31;9(8):812. doi: 10.3390/jof9080812. J Fungi (Basel). 2023. PMID: 37623583 Free PMC article. Review.
References
-
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte G, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1 , RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1. ADAR, and IFIH1. Am J Med Genet Part A (2015) 167A(2):296–312. doi: 10.1002/ajmg.a.36887 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous