

Using whole genome sequence to compare variant callers and breed differences of US sheep
- PMID: 36685812
- PMCID: PMC9846548
- DOI: 10.3389/fgene.2022.1060882
Using whole genome sequence to compare variant callers and breed differences of US sheep
Abstract
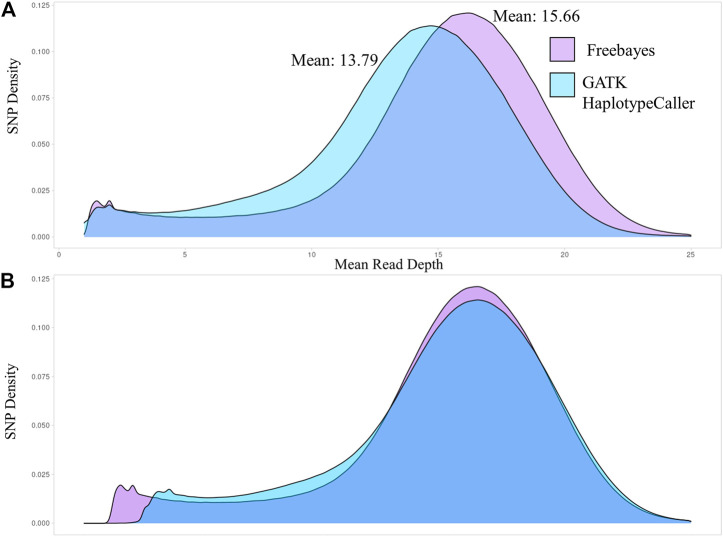
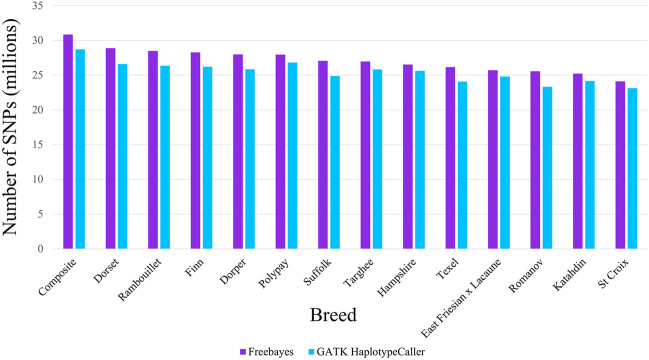
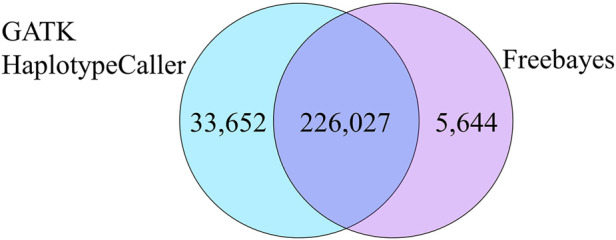
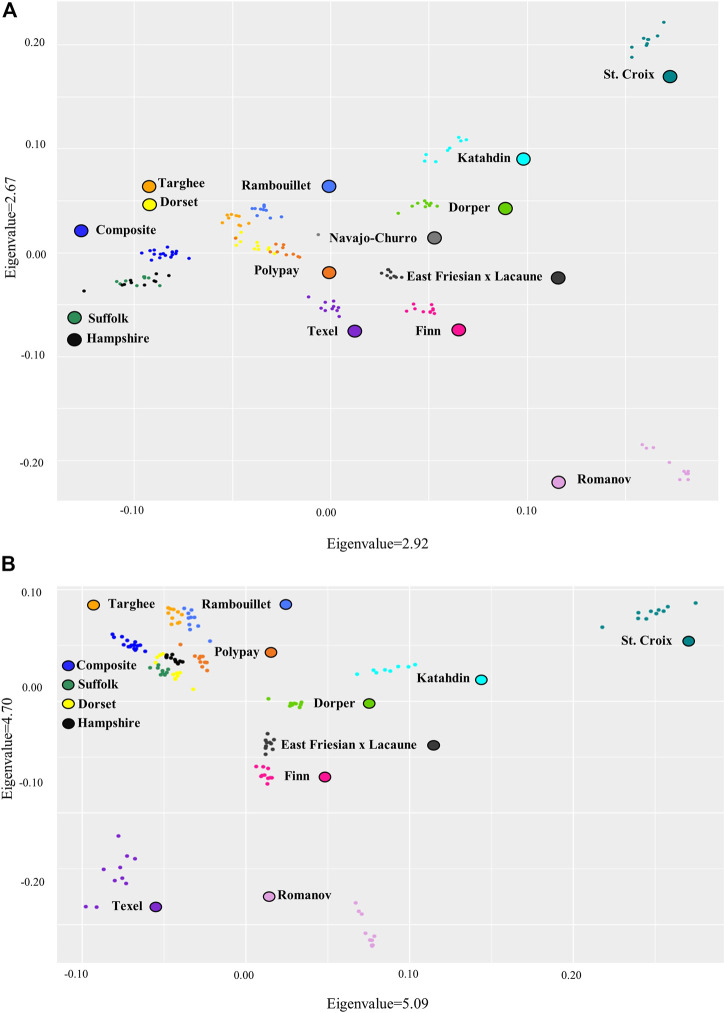
As whole genome sequence (WGS) data sets have become abundant and widely available, so has the need for variant detection and scoring. The aim of this study was to compare the accuracy of commonly used variant calling programs, Freebayes and GATK HaplotypeCaller (GATK-HC), and to use U.S. sheep WGS data sets to identify novel breed-associated SNPs. Sequence data from 145 sheep consisting of 14 U.S. breeds were filtered and biallelic single nucleotide polymorphisms (SNPs) were retained for genotyping analyses. Genotypes from both programs were compared to each other and to genotypes from bead arrays. The SNPs from WGS were compared to the bead array data with breed heterozygosity, principal component analysis and identifying breed associated SNPs to analyze genetic diversity. The average sequence read depth was 2.78 reads greater with 6.11% more SNPs being identified in Freebayes compared to GATK-HC. The genotype concordance of the variant callers to bead array data was 96.0% and 95.5% for Freebayes and GATK-HC, respectively. Genotyping with WGS identified 10.5 million SNPs from all 145 sheep. This resulted in an 8% increase in measured heterozygosity and greater breed separation in the principal component analysis compared to the bead array analysis. There were 1,849 SNPs identified in only the Romanov sheep where all 10 rams were homozygous for one allele and the remaining 135 sheep from 13 breeds were homozygous for the opposite allele. Both variant calling programs had greater than 95% concordance of SNPs with bead array data, and either was suitably accurate for ovine WGS data sets. The use of WGS SNPs improved the resolution of PCA analysis and was critical for identifying Romanov breed-associated SNPs. Subsets of such SNPs could be used to estimate germplasm composition in animals without pedigree information.
Keywords: GATK HaplotypeCaller (HC); freebayes; sheep; variant callers; whole genome sequence.
Copyright © 2023 Stegemiller, Redden, Notter, Taylor, Taylor, Cockett, Heaton, Kalbfleisch and Murdoch.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Comparison among three variant callers and assessment of the accuracy of imputation from SNP array data to whole-genome sequence level in chicken.BMC Genomics. 2015 Oct 21;16:824. doi: 10.1186/s12864-015-2059-2. BMC Genomics. 2015. PMID: 26486989 Free PMC article.
-
Evaluation of variant calling tools for large plant genome re-sequencing.BMC Bioinformatics. 2020 Aug 17;21(1):360. doi: 10.1186/s12859-020-03704-1. BMC Bioinformatics. 2020. PMID: 32807073 Free PMC article.
-
Assessing single nucleotide variant detection and genotype calling on whole-genome sequenced individuals.Bioinformatics. 2014 Jun 15;30(12):1707-13. doi: 10.1093/bioinformatics/btu067. Epub 2014 Feb 19. Bioinformatics. 2014. PMID: 24558117
-
Detailed comparison of two popular variant calling packages for exome and targeted exon studies.PeerJ. 2014 Sep 30;2:e600. doi: 10.7717/peerj.600. eCollection 2014. PeerJ. 2014. PMID: 25289185 Free PMC article.
-
Single Nucleotide Polymorphism Identification in Polyploids: A Review, Example, and Recommendations.Mol Plant. 2015 Jun;8(6):831-46. doi: 10.1016/j.molp.2015.02.002. Epub 2015 Feb 10. Mol Plant. 2015. PMID: 25676455 Review.
Cited by
-
Performance analysis of conventional and AI-based variant callers using short and long reads.BMC Bioinformatics. 2023 Dec 14;24(1):472. doi: 10.1186/s12859-023-05596-3. BMC Bioinformatics. 2023. PMID: 38097928 Free PMC article.
-
Identifying Genetic Predisposition to Dozer Lamb Syndrome: A Semi-Lethal Muscle Weakness Disease in Sheep.Genes (Basel). 2025 Jan 14;16(1):83. doi: 10.3390/genes16010083. Genes (Basel). 2025. PMID: 39858630 Free PMC article.
-
Species-specific dynamics may cause deviations from general biogeographical predictions - evidence from a population genomics study of a New Guinean endemic passerine bird family (Melampittidae).PLoS One. 2024 May 23;19(5):e0293715. doi: 10.1371/journal.pone.0293715. eCollection 2024. PLoS One. 2024. PMID: 38781204 Free PMC article.
-
Molecular Cytogenetics in Domestic Bovids: A Review.Animals (Basel). 2023 Mar 6;13(5):944. doi: 10.3390/ani13050944. Animals (Basel). 2023. PMID: 36899801 Free PMC article. Review.
References
-
- Berry D.P., O’Brien A., Wall E., McDerott K., Randles S., Flynn P., Park S., Grose J., Weld R., McHugh N. (2016). Inter- and intra-reproducibility of genotypes from sheep technical replicates on Illumina and Affymetrix platforms. Genet. Sel. Evol. 48 (86), 86. 10.1186/s12711-016-0267-0 - DOI - PMC - PubMed
-
- Fan B., Du Z.Q., Gorbach D.M., Rothschild M.F. (2010). Development and Application of high-density SNP Arrays in Genomic Studies of Domestic Animals. Asian-Australas. J. Anim. Sci. 23 (7), 833–847. 10.5713/ajas.2010.r.03 - DOI
LinkOut - more resources
Full Text Sources
Research Materials