

Ribosomal RNA (rRNA) sequences from 33 globally distributed mosquito species for improved metagenomics and species identification
- PMID: 36688360
- PMCID: PMC10014081
- DOI: 10.7554/eLife.82762
Ribosomal RNA (rRNA) sequences from 33 globally distributed mosquito species for improved metagenomics and species identification
Abstract
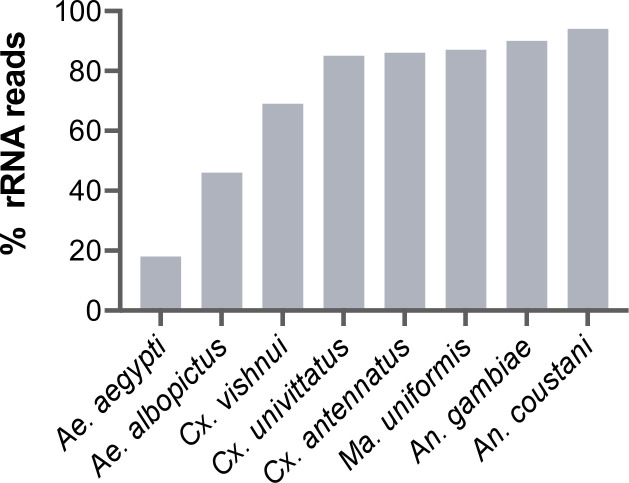
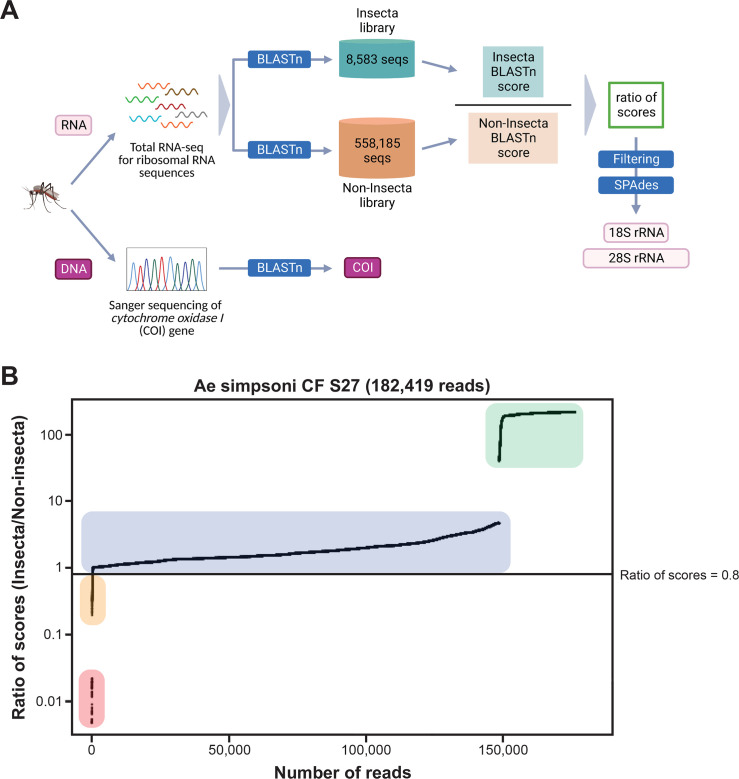
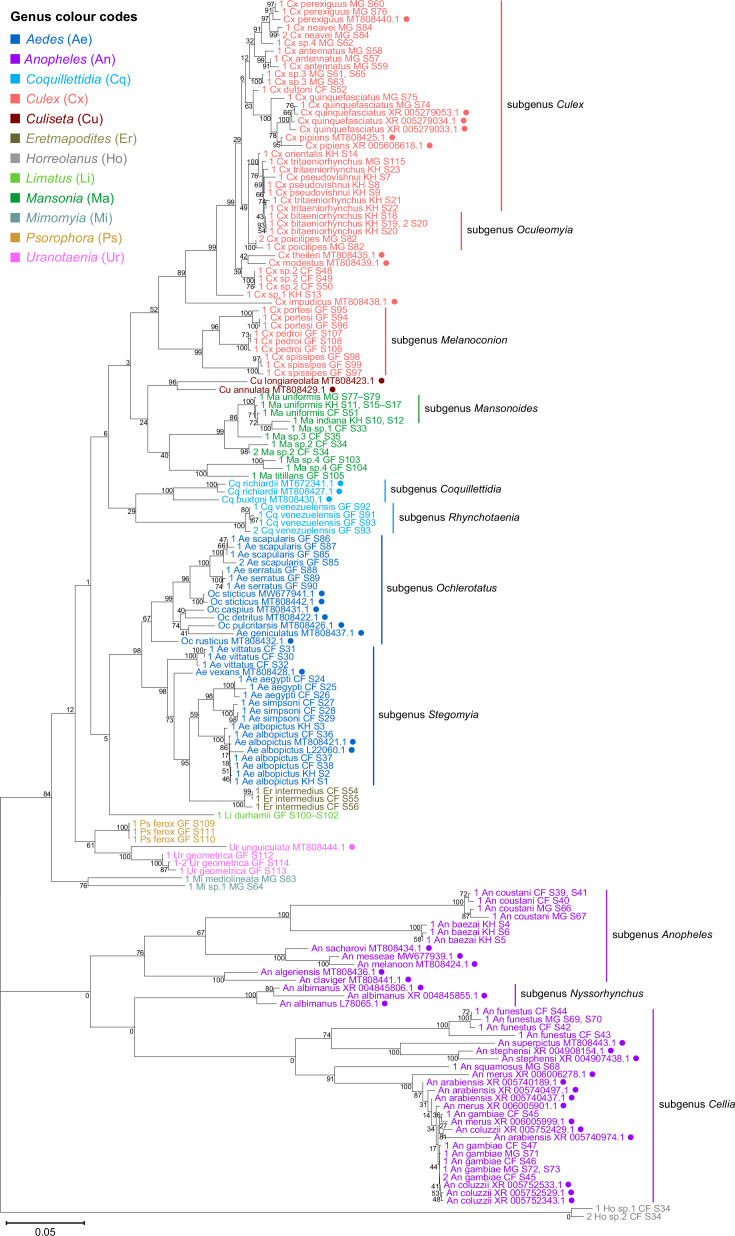
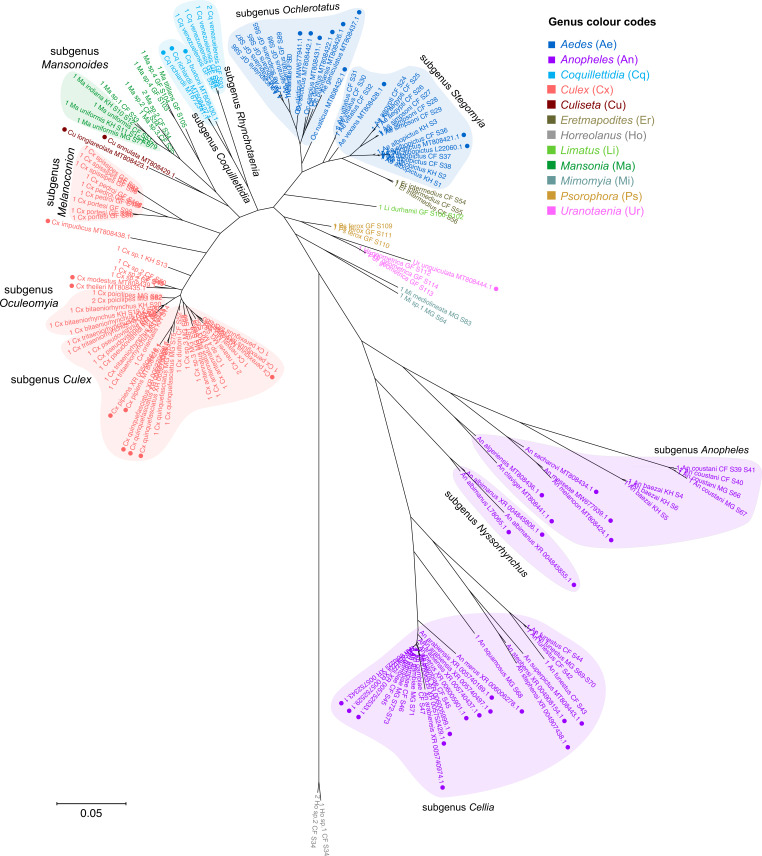
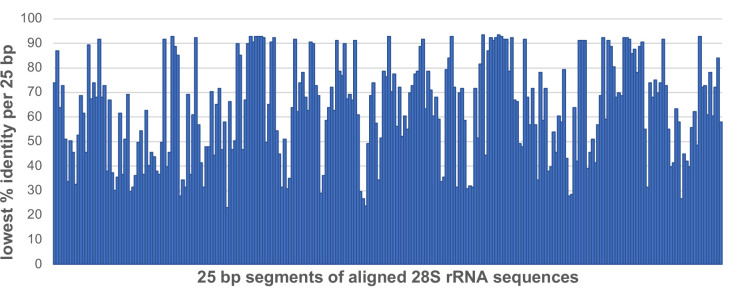
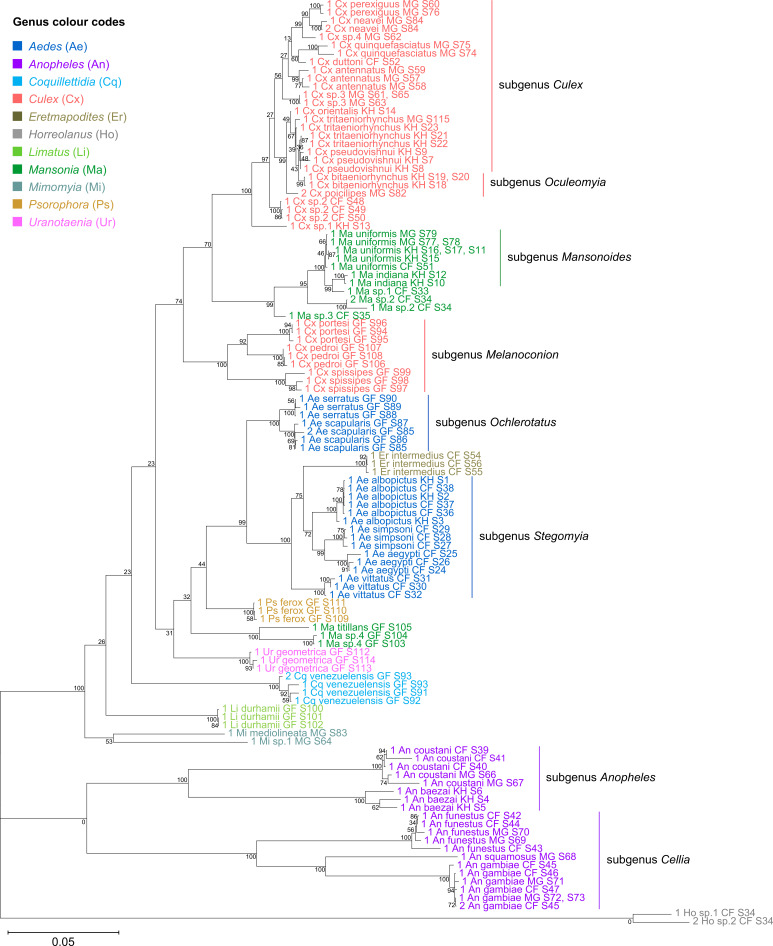
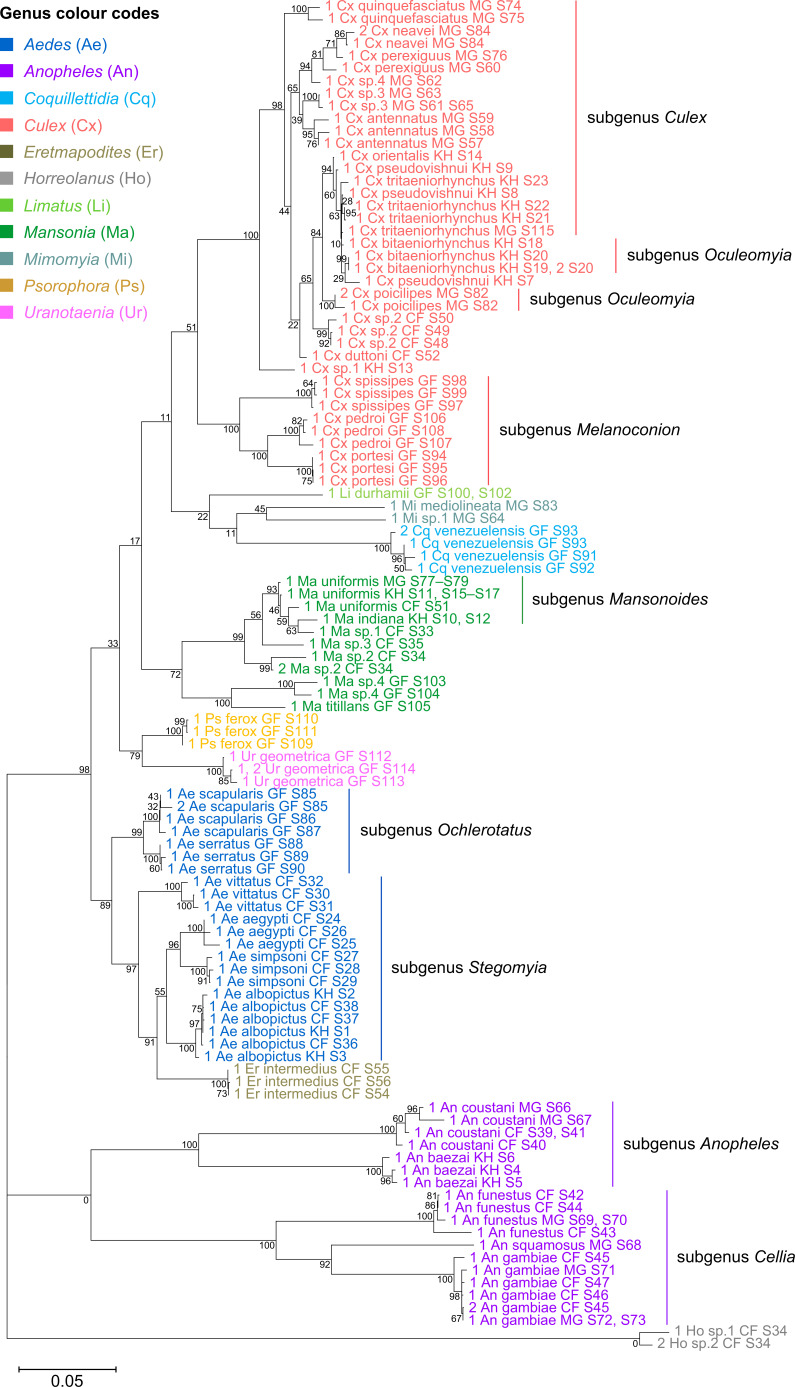
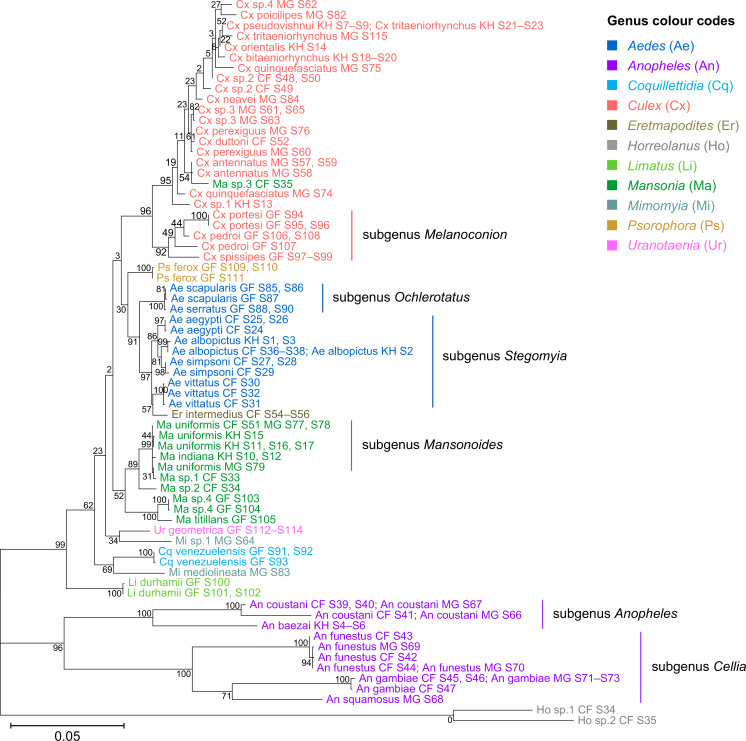
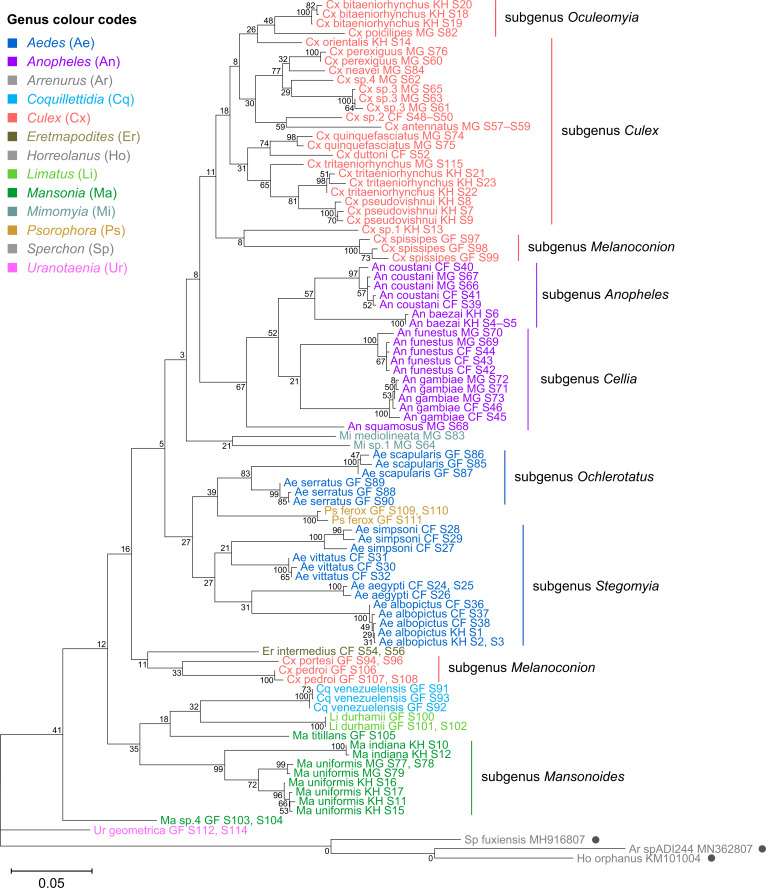
Total RNA sequencing (RNA-seq) is an important tool in the study of mosquitoes and the RNA viruses they vector as it allows assessment of both host and viral RNA in specimens. However, there are two main constraints. First, as with many other species, abundant mosquito ribosomal RNA (rRNA) serves as the predominant template from which sequences are generated, meaning that the desired host and viral templates are sequenced far less. Second, mosquito specimens captured in the field must be correctly identified, in some cases to the sub-species level. Here, we generate mosquito rRNA datasets which will substantially mitigate both of these problems. We describe a strategy to assemble novel rRNA sequences from mosquito specimens and produce an unprecedented dataset of 234 full-length 28S and 18S rRNA sequences of 33 medically important species from countries with known histories of mosquito-borne virus circulation (Cambodia, the Central African Republic, Madagascar, and French Guiana). These sequences will allow both physical and computational removal of rRNA from specimens during RNA-seq protocols. We also assess the utility of rRNA sequences for molecular taxonomy and compare phylogenies constructed using rRNA sequences versus those created using the gold standard for molecular species identification of specimens-the mitochondrial cytochrome c oxidase I (COI) gene. We find that rRNA- and COI-derived phylogenetic trees are incongruent and that 28S and concatenated 28S+18S rRNA phylogenies reflect evolutionary relationships that are more aligned with contemporary mosquito systematics. This significant expansion to the current rRNA reference library for mosquitoes will improve mosquito RNA-seq metagenomics by permitting the optimization of species-specific rRNA depletion protocols for a broader range of species and streamlining species identification by rRNA sequence and phylogenetics.
Keywords: RNA-seq; infectious disease; metagenomics; microbiology; molecular taxonomy; mosquito; ribosomal RNA; surveillance.
© 2023, Koh et al.
Conflict of interest statement
CK, LF, HB, CN, SB, PD, NG, RG, JD, MS No competing interests declared
Figures
Update of
- doi: 10.1101/2022.02.01.478639
Similar articles
-
Barcoding of Italian mosquitoes (BITMO): generation and validation of DNA barcoding reference libraries for native and alien species of Culicidae.Parasit Vectors. 2024 Sep 28;17(1):407. doi: 10.1186/s13071-024-06478-0. Parasit Vectors. 2024. PMID: 39342262 Free PMC article.
-
28S rRNA sequences for Linguatula spp.Parasitol Res. 2022 Jun;121(6):1799-1804. doi: 10.1007/s00436-022-07507-6. Epub 2022 Apr 1. Parasitol Res. 2022. PMID: 35362745 Free PMC article.
-
Phylogenetic analysis of the spider mite sub-family Tetranychinae (Acari: Tetranychidae) based on the mitochondrial COI gene and the 18S and the 5' end of the 28S rRNA genes indicates that several genera are polyphyletic.PLoS One. 2014 Oct 7;9(10):e108672. doi: 10.1371/journal.pone.0108672. eCollection 2014. PLoS One. 2014. PMID: 25289639 Free PMC article.
-
Molecular differentiation of Sarcocystis buffalonis and Sarcocystis levinei in water buffaloes (Bubalus bubalis) from Sarcocystis hirsuta and Sarcocystis cruzi in cattle (Bos taurus).Parasitol Res. 2016 Jun;115(6):2459-71. doi: 10.1007/s00436-016-4998-1. Epub 2016 Mar 16. Parasitol Res. 2016. PMID: 26979729
-
Translating mosquito viromes into vector management strategies.Trends Parasitol. 2024 Jan;40(1):10-20. doi: 10.1016/j.pt.2023.11.002. Epub 2023 Dec 7. Trends Parasitol. 2024. PMID: 38065789 Review.
Cited by
-
Genetic diversity and Wolbachia infection in the Japanese encephalitis virus vector Culex tritaeniorhynchus in the Republic of Korea.Parasit Vectors. 2024 Dec 18;17(1):518. doi: 10.1186/s13071-024-06595-w. Parasit Vectors. 2024. PMID: 39695811 Free PMC article.
-
The complete mitochondrial genome of Culex hutchinsoni (Diptera: Culicidae) and its phylogenetic analysis.Mitochondrial DNA B Resour. 2024 Nov 27;9(11):1615-1619. doi: 10.1080/23802359.2024.2432350. eCollection 2024. Mitochondrial DNA B Resour. 2024. PMID: 39624599 Free PMC article.
-
The application of metagenomics in the detection of arboviruses in mosquitoes (Diptera: Culicidade): a systematic review.Rev Inst Med Trop Sao Paulo. 2025 Jul 7;67:e46. doi: 10.1590/S1678-9946202567046. eCollection 2025. Rev Inst Med Trop Sao Paulo. 2025. PMID: 40638514 Free PMC article.
References
-
- Aspen S, Savage HM. Polymerase chain reaction assay identifies North American members of the Culex pipiens complex based on nucleotide sequence differences in the acetylcholinesterase gene Ace.2. Journal of the American Mosquito Control Association. 2003;19:323–328. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
