

Eliminating oncogenic RAS: back to the future at the drawing board
- PMID: 36688434
- PMCID: PMC9987992
- DOI: 10.1042/BST20221343
Eliminating oncogenic RAS: back to the future at the drawing board
Abstract
RAS drug development has made enormous strides in the past ten years, with the first direct KRAS inhibitor being approved in 2021. However, despite the clinical success of covalent KRAS-G12C inhibitors, we are immediately confronted with resistances as commonly found with targeted drugs. Previously believed to be undruggable due to its lack of obvious druggable pockets, a couple of new approaches to hit this much feared oncogene have now been carved out. We here concisely review these approaches to directly target four druggable sites of RAS from various angles. Our analysis focuses on the lessons learnt during the development of allele-specific covalent and non-covalent RAS inhibitors, the potential of macromolecular binders to facilitate the discovery and validation of targetable sites on RAS and finally an outlook on a future that may engage more small molecule binders to become drugs. We foresee that the latter could happen mainly in two ways: First, non-covalent small molecule inhibitors may be derived from the development of covalent binders. Second, reversible small molecule binders could be utilized for novel targeting modalities, such as degraders of RAS. Provided that degraders eliminate RAS by recruiting differentially expressed E3-ligases, this approach could enable unprecedented tissue- or developmental stage-specific destruction of RAS with potential advantages for on-target toxicity. We conclude that novel creative ideas continue to be important to exterminate RAS in cancer and other RAS pathway-driven diseases, such as RASopathies.
Keywords: RAS; cancer; drug development.
© 2023 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
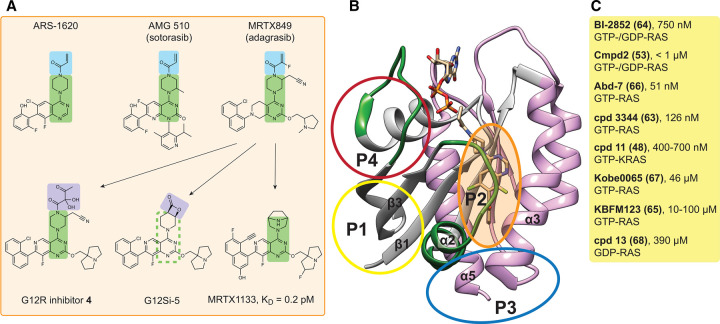
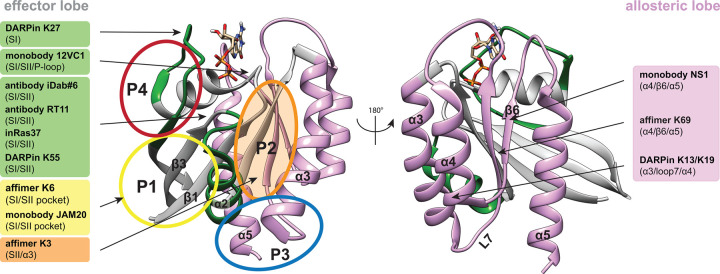
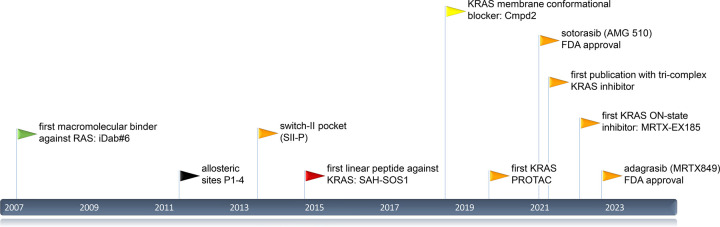
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous