

Parkin coregulates glutathione metabolism in adult mammalian brain
- PMID: 36691076
- PMCID: PMC9869535
- DOI: 10.1186/s40478-022-01488-4
Parkin coregulates glutathione metabolism in adult mammalian brain
Abstract
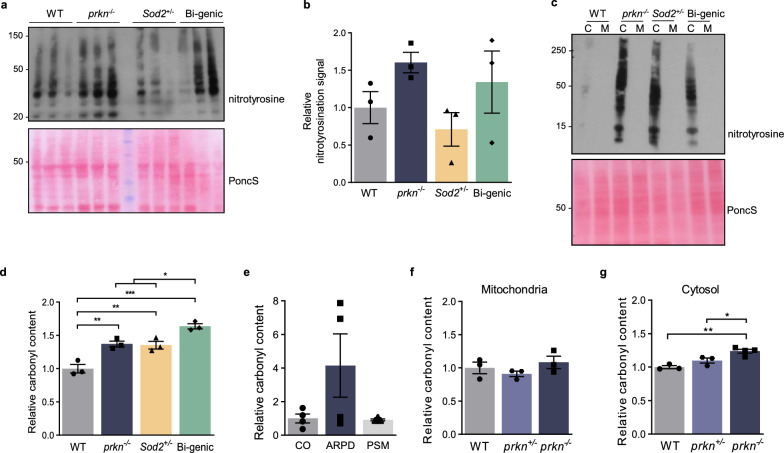
We recently discovered that the expression of PRKN, a young-onset Parkinson disease-linked gene, confers redox homeostasis. To further examine the protective effects of parkin in an oxidative stress model, we first combined the loss of prkn with Sod2 haploinsufficiency in mice. Although adult prkn-/-//Sod2± animals did not develop dopamine cell loss in the S. nigra, they had more reactive oxidative species and a higher concentration of carbonylated proteins in the brain; bi-genic mice also showed a trend for more nitrotyrosinated proteins. Because these redox changes were seen in the cytosol rather than mitochondria, we next explored the thiol network in the context of PRKN expression. We detected a parkin deficiency-associated increase in the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) in murine brain, PRKN-linked human cortex and several cell models. This shift resulted from enhanced recycling of GSSG back to GSH via upregulated glutathione reductase activity; it also correlated with altered activities of redox-sensitive enzymes in mitochondria isolated from mouse brain (e.g., aconitase-2; creatine kinase). Intriguingly, human parkin itself showed glutathione-recycling activity in vitro and in cells: For each GSSG dipeptide encountered, parkin regenerated one GSH molecule and was S-glutathionylated by the other (GSSG + P-SH [Formula: see text] GSH + P-S-SG), including at cysteines 59, 95 and 377. Moreover, parkin's S-glutathionylation was reversible by glutaredoxin activity. In summary, we found that PRKN gene expression contributes to the network of available thiols in the cell, including by parkin's participation in glutathione recycling, which involves a reversible, posttranslational modification at select cysteines. Further, parkin's impact on redox homeostasis in the cytosol can affect enzyme activities elsewhere, such as in mitochondria. We posit that antioxidant functions of parkin may explain many of its previously described, protective effects in vertebrates and invertebrates that are unrelated to E3 ligase activity.
Keywords: Early-onset Parkinson disease; Glutathione metabolism; Mass spectrometry; Parkin; Posttranslational modification; Prkn; Redox stress; Sod2.
© 2023. The Author(s).
Conflict of interest statement
The Ottawa Hospital receives payments from BioLegend Inc. related to licensing agreements for immunological reagents related to parkin and α-synuclein. Dr. M. Schlossmacher received travel reimbursements from the Michael J. Fox Foundation for Parkinson’s Research for participation in industry summits and consulting fees (Biogen; Neuramedy; Samsara) as well as royalties from Eli Lilly for patents unrelated to this work. Dr. A. Holmgren (deceased) served as chairman and senior scientist at IMCO Corporation Ltd AB, Stockholm, Sweden. No additional, potentially competing financial interests are declared.
Figures





References
-
- Abela L, Spiegel R, Crowther LM, Klein A, Steindl K, Papuc SM, Joset P, Zehavi Y, Rauch A, Plecko B, Simmons TL. Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency. PLoS ONE. 2017;12:e0176363. doi: 10.1371/journal.pone.0176363. - DOI - PMC - PubMed
-
- Adedara AO, Babalola AD, Stephano F, Awogbindin IO, Olopade JO, Rocha JBT, Whitworth AJ, Abolaji AO. An assessment of the rescue action of resveratrol in parkin loss of function-induced oxidative stress in Drosophila melanogaster. Sci Rep. 2022;12:3922. doi: 10.1038/s41598-022-07909-7. - DOI - PMC - PubMed
-
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Kruger R, Surmeier DJ, Krainc D. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017;357:1255–1261. doi: 10.1126/science.aam9080. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
