

Chronic alcohol metabolism results in DNA repair infidelity and cell cycle-induced senescence in neurons
- PMID: 36691110
- PMCID: PMC9924945
- DOI: 10.1111/acel.13772
Chronic alcohol metabolism results in DNA repair infidelity and cell cycle-induced senescence in neurons
Abstract
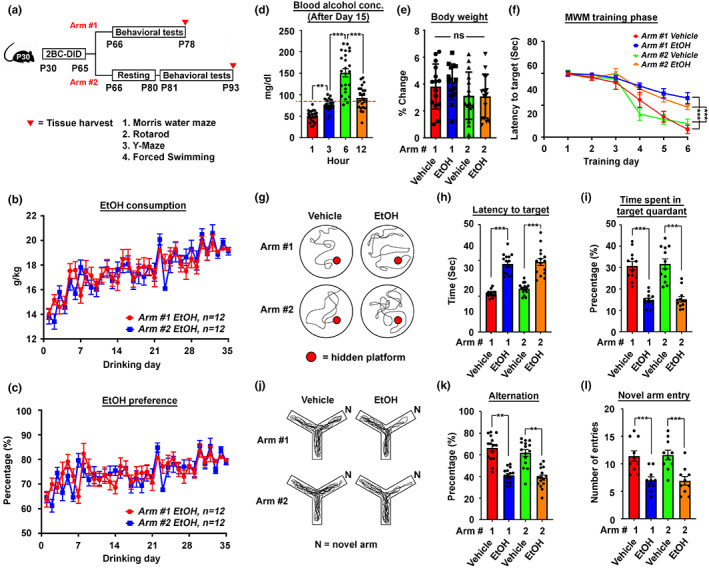
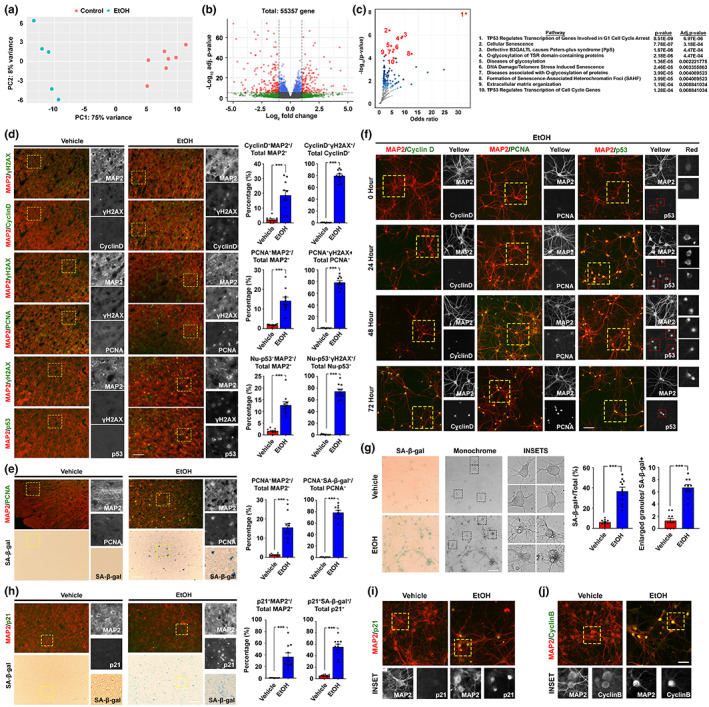
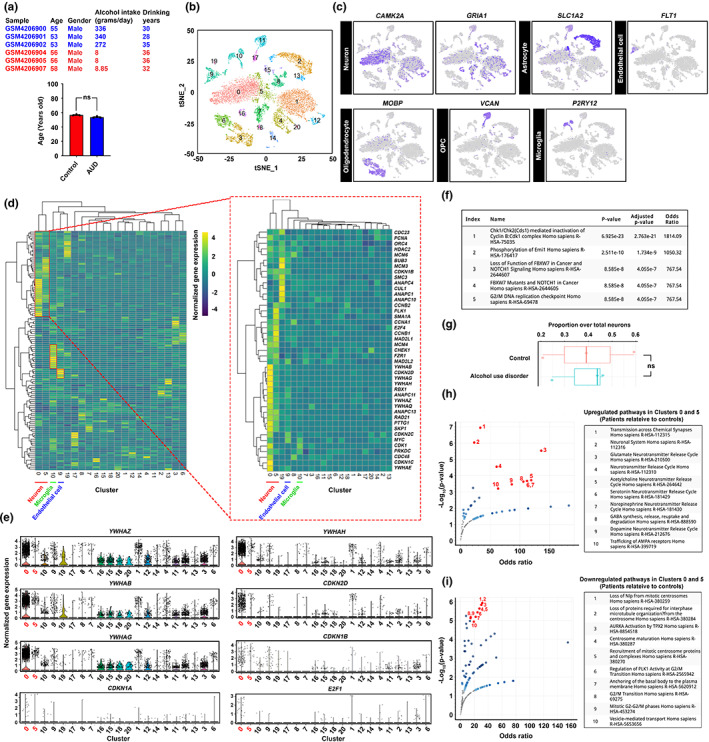
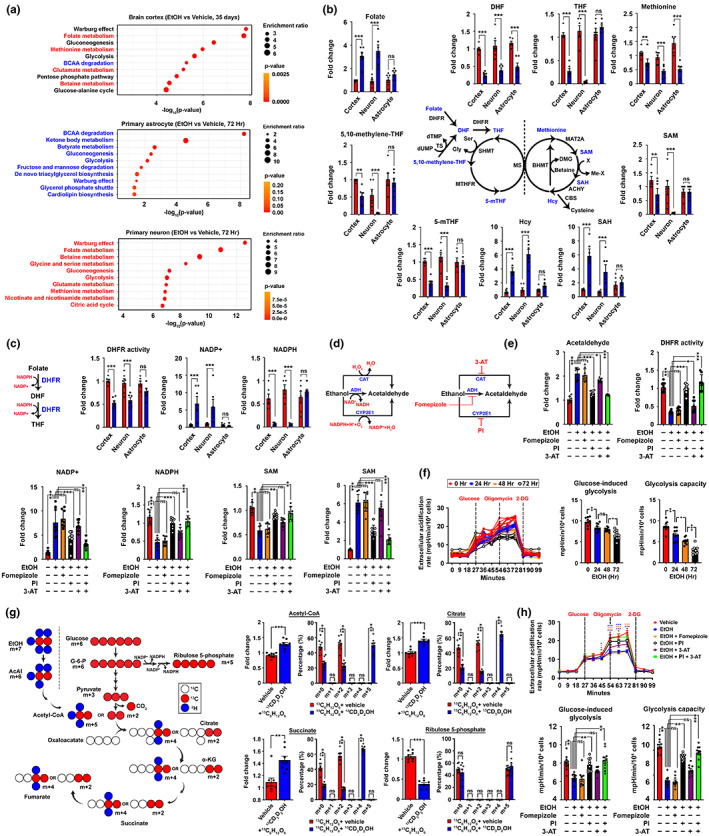
Chronic binge-like drinking is a risk factor for age-related dementia, however, the lasting and irreversible effect of alcohol on the brain remains elusive. Transcriptomic changes in brain cortices revealed pro-ageing hallmarks upon chronic ethanol exposure and these changes predominantly occur in neurons. The changes are attributed to a prioritized ethyl alcohol oxidation in these cells via the NADPH-dependent cytochrome pathway. This hijacks the folate metabolism of the 1-carbon network which supports the pathway choice of DNA repair via the non-cell cycle-dependent mismatch repair networks. The lost-in-function of such results in the de-inactivation of the less preferred cell cycle-dependent homologous recombination (HR) repair, forcing these post-mitotic cells to re-engage in a cell cycle-like process. However, mature neurons are post-mitotic. Therefore, instead of successfully completing a full round of cell cycle which is necessary for the completion of HR-mediated repair; these cells are arrested at checkpoints. The resulting persistence of repair intermediates induces and promotes the nuclear accumulation of p21 and cyclin B-a trigger for permanent cell cycle exits and irreversible senescence response. Supplementation of bioactive 5-methyl tetrahydrofolate simultaneously at times with ethyl alcohol exposure supports the fidelity of the 1-carbon network and hence the activity of the mismatch repair. This prevents aberrant and irreversible cell cycle re-entry and senescence events of neurons. Together, our findings offer a direct connection between binge-drinking behaviour and its irreversible impact on the brain, which makes it a potential risk factor for dementia.
Keywords: 1-carbon metabolism; DNA damage response; cell cycle re-entry; chronic alcohol use; metabolic reprogramming; neuronal senescence.
© 2023 The Authors. Aging Cell published by Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
None.
Figures
Similar articles
-
Benzo[a]pyrene represses DNA repair through altered E2F1/E2F4 function marking an early event in DNA damage-induced cellular senescence.Nucleic Acids Res. 2020 Dec 2;48(21):12085-12101. doi: 10.1093/nar/gkaa965. Nucleic Acids Res. 2020. PMID: 33166399 Free PMC article.
-
DNA Damage Response-Associated Cell Cycle Re-Entry and Neuronal Senescence in Brain Aging and Alzheimer's Disease.J Alzheimers Dis. 2023;94(s1):S429-S451. doi: 10.3233/JAD-220203. J Alzheimers Dis. 2023. PMID: 35848025 Free PMC article. Review.
-
CD95 ligand induces senescence in mismatch repair-deficient human colon cancer via chronic caspase-mediated induction of DNA damage.Cell Death Dis. 2017 Mar 16;8(3):e2669. doi: 10.1038/cddis.2017.87. Cell Death Dis. 2017. PMID: 28300842 Free PMC article.
-
Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-κB.Cancer Res. 2019 Jan 1;79(1):99-113. doi: 10.1158/0008-5472.CAN-18-1733. Epub 2018 Oct 25. Cancer Res. 2019. PMID: 30361254
-
DNA-damage response network at the crossroads of cell-cycle checkpoints, cellular senescence and apoptosis.J Zhejiang Univ Sci B. 2007 Jun;8(6):377-97. doi: 10.1631/jzus.2007.B0377. J Zhejiang Univ Sci B. 2007. PMID: 17565509 Free PMC article. Review.
Cited by
-
Senescent brain cell types in Alzheimer's disease: Pathological mechanisms and therapeutic opportunities.Neurotherapeutics. 2025 Apr;22(3):e00519. doi: 10.1016/j.neurot.2024.e00519. Epub 2025 Jan 6. Neurotherapeutics. 2025. PMID: 39765417 Free PMC article. Review.
-
Nature's Secret Neuro-Regeneration Pathway in Axolotls, Polychaetes and Planarians for Human Therapeutic Target Pathways.Int J Mol Sci. 2024 Nov 6;25(22):11904. doi: 10.3390/ijms252211904. Int J Mol Sci. 2024. PMID: 39595973 Free PMC article. Review.
-
The role of cellular senescence in neurodegenerative diseases.Arch Toxicol. 2024 Aug;98(8):2393-2408. doi: 10.1007/s00204-024-03768-5. Epub 2024 May 15. Arch Toxicol. 2024. PMID: 38744709 Free PMC article. Review.
-
Recent advances in alcohol metabolism: from the gut to the brain.Physiol Rev. 2025 Oct 1;105(4):2501-2535. doi: 10.1152/physrev.00053.2024. Epub 2025 Jul 10. Physiol Rev. 2025. PMID: 40637545 Free PMC article. Review.
-
Neuronal cell cycle reentry events in the aging brain are more prevalent in neurodegeneration and lead to cellular senescence.PLoS Biol. 2024 Apr 23;22(4):e3002559. doi: 10.1371/journal.pbio.3002559. eCollection 2024 Apr. PLoS Biol. 2024. PMID: 38652714 Free PMC article.
References
-
- Abdel‐Azeim, S. , Li, X. , Chung, L. W. , & Morokuma, K. (2011). Zinc‐homocysteine binding in cobalamin‐dependent methionine synthase and its role in the substrate activation: DFT, ONIOM, and QM/MM molecular dynamics studies. Journal of Computational Chemistry, 32(15), 3154–3167. 10.1002/jcc.21895 - DOI - PubMed
-
- Brunet, A. , Kanai, F. , Stehn, J. , Xu, J. , Sarbassova, D. , Frangioni, J. V. , Dalal, S. N. , Decaprio, J. A. , Greenberg, M. E. , & Yaffe, M. B. (2002). 14‐3‐3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. The Journal of Cell Biology, 156(5), 817–828. 10.1083/jcb.200112059 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
