

Predominant Liver Cystic Disease in a New Heterozygotic PKHD1 Variant: A Case Report
- PMID: 36691356
- PMCID: PMC9883601
- DOI: 10.12659/AJCR.938507
Predominant Liver Cystic Disease in a New Heterozygotic PKHD1 Variant: A Case Report
Abstract
BACKGROUND The polycystic kidney and hepatic disease 1 (PKHD1) gene codes for fibrocystin-polyductin, a protein that takes part in cell-signaling for cell differentiation, especially in kidney tubules and bile ducts. A homozygous or compound heterozygous defect in this gene can cause autosomal recessive polycystic kidney disease (ARPKD). Polycystic liver disease (PCLD) can also be caused by single heterozygous variants in the PKHD1 gene. ARPKD presents with renal insufficiency and cystic dilatation of bile ducts, although disease is not expected with a single heterozygous mutation. PCLD presents with multiple cysts in the liver and dilated bile ducts as well, but with less of an impact on the kidneys than with ARPKD. Our purpose in publishing this report is to introduce an as-yet unknown variant to the body of genetic defects associated with ARPKD and PCLD, as well as to argue for the likely pathogenicity of the variant according to the prevailing criteria used for classifying gene variants. CASE REPORT We present a patient with a de novo PKHD1 variant currently classified as a variant of unknown significance manifesting with bilaterally enlarged cystic kidneys and echogenic cystic structures in the hepatic portal system, indicative of cystic disease. CONCLUSIONS Given this patient's liver and kidney presentation that does not fully align with either ARPKD or PCLD, the authors believe that the single heterozygous variant in this patient's PKHD1 gene is worthy of reporting. This new single heterozygous variant in PKHD1 gene causing cystic kidney and cystic hepatic disease in the patient should be considered 'likely pathogenic' according to the criteria set by the American College of Medical Genetics.
Conflict of interest statement
Figures
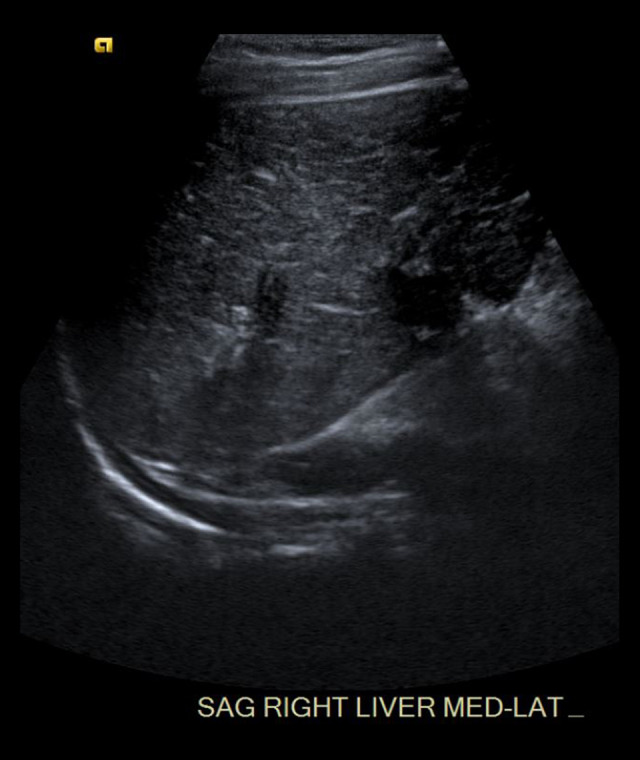
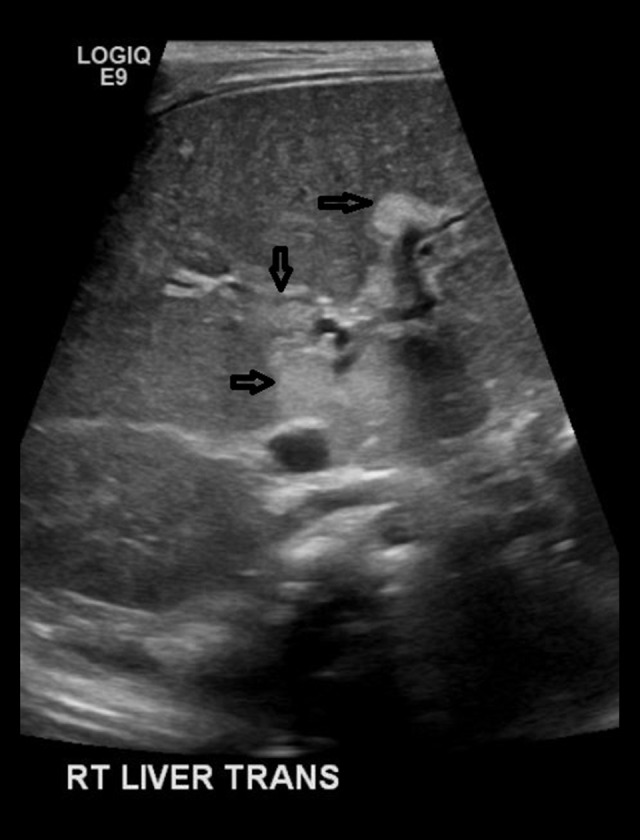
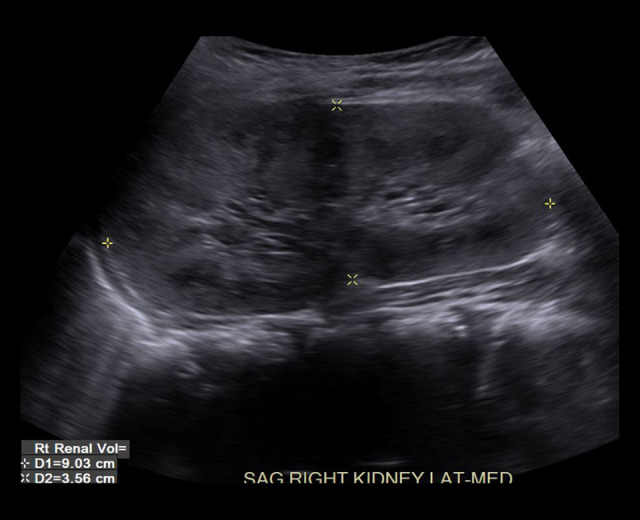
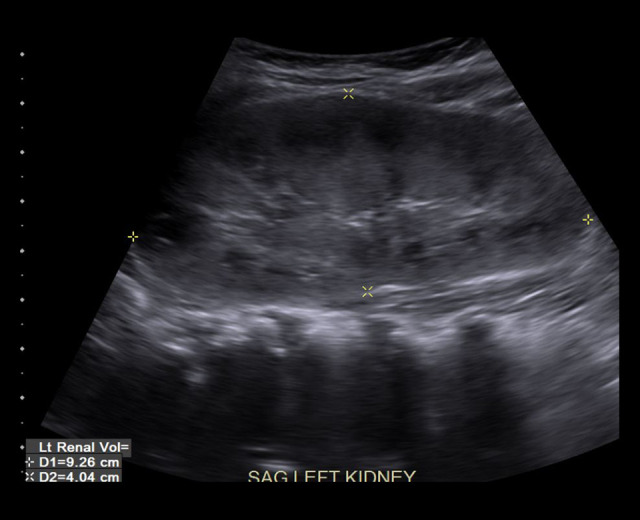
Similar articles
-
Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease.Pediatr Nephrol. 2008 May;23(5):733-41. doi: 10.1007/s00467-007-0735-4. Epub 2008 Feb 20. Pediatr Nephrol. 2008. PMID: 18286309
-
Autosomal Recessive Polycystic Kidney Disease – PKHD1.2001 Jul 19 [updated 2024 Apr 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2001 Jul 19 [updated 2024 Apr 4]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301501 Free Books & Documents. Review.
-
Pkhd1cyli/cyli mice have altered renal Pkhd1 mRNA processing and hormonally sensitive liver disease.J Mol Med (Berl). 2023 Sep;101(9):1141-1151. doi: 10.1007/s00109-023-02351-2. Epub 2023 Aug 16. J Mol Med (Berl). 2023. PMID: 37584738 Free PMC article.
-
Boy with autosomal recessive polycystic kidney and autosomal dominant polycystic liver disease.Pediatr Nephrol. 2012 Jul;27(7):1197-200. doi: 10.1007/s00467-012-2137-5. Epub 2012 Mar 14. Pediatr Nephrol. 2012. PMID: 22415584
-
Imaging manifestations of Caroli disease with autosomal recessive polycystic kidney disease: a case report and literature review.BMC Pregnancy Childbirth. 2021 Apr 12;21(1):294. doi: 10.1186/s12884-021-03768-8. BMC Pregnancy Childbirth. 2021. PMID: 33845788 Free PMC article. Review.
Cited by
-
Pathogenic relationship between phenotypes of ARPKD and novel compound heterozygous mutations of PKHD1.Front Genet. 2024 Jul 2;15:1429336. doi: 10.3389/fgene.2024.1429336. eCollection 2024. Front Genet. 2024. PMID: 39015774 Free PMC article.
-
Co-Occurrence of Neurofibromatosis Type 1 and Polycystic Liver Disease: A Case of Hypertension with PKHD1 Variant.Am J Case Rep. 2025 Apr 5;26:e947141. doi: 10.12659/AJCR.947141. Am J Case Rep. 2025. PMID: 40186343 Free PMC article.
References
-
- Zerres K, Rudnik-Schoneborn S, Steinkamm C, et al. Autosomal recessive polycystic kidney disease. J Mol Med. 1998;76:303–9. - PubMed
-
- Sanyal AJ, Boyer TD, Terrault N, Lindor KD, editors. Zakim and Boyer’s Hepatology: A Textbook of Liver Disease. Seventh edition. Pennsylvania: Elsevier; 2018.
-
- Sweeney WE, Avner ED. Polycystic kidney disease, autosomal recessive. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2001. Jul 19, pp. 1993–2022. [Updated 2019 Feb 14]. In: Available from: https://www.ncbi.nlm.nih.gov/books/NBK1326/ - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
