

Mouse models of fragile X-related disorders
- PMID: 36692473
- PMCID: PMC9903145
- DOI: 10.1242/dmm.049485
Mouse models of fragile X-related disorders
Abstract
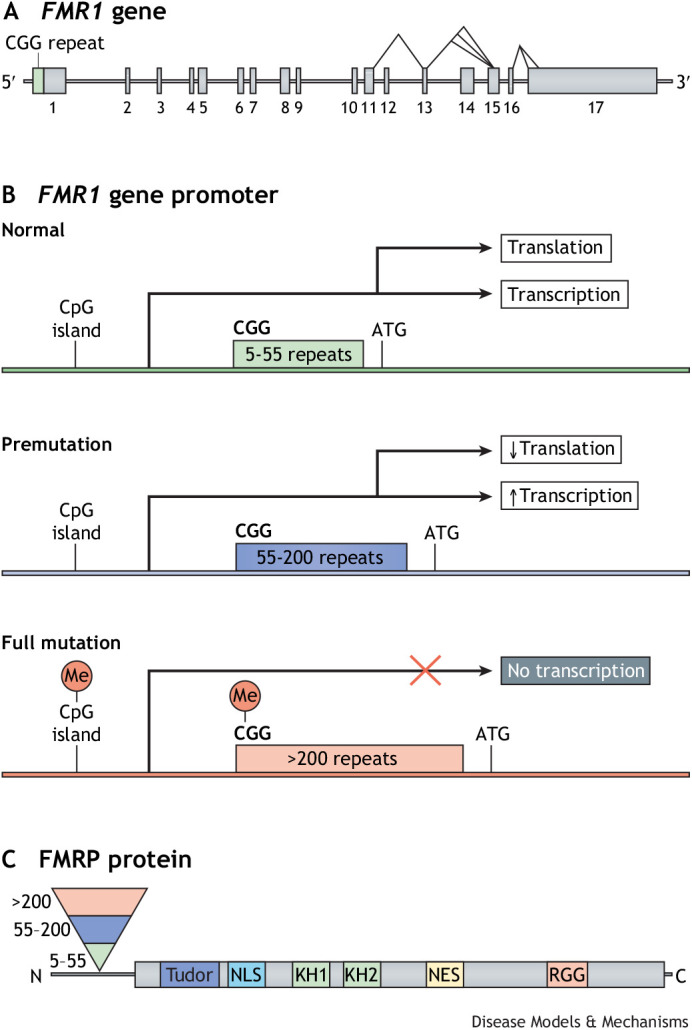
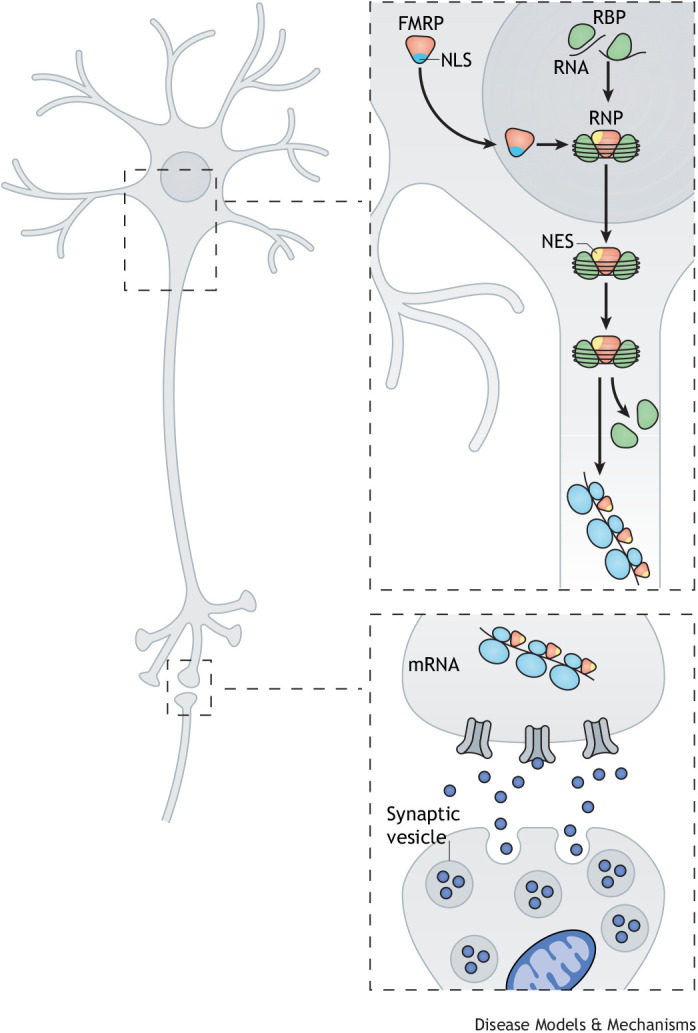
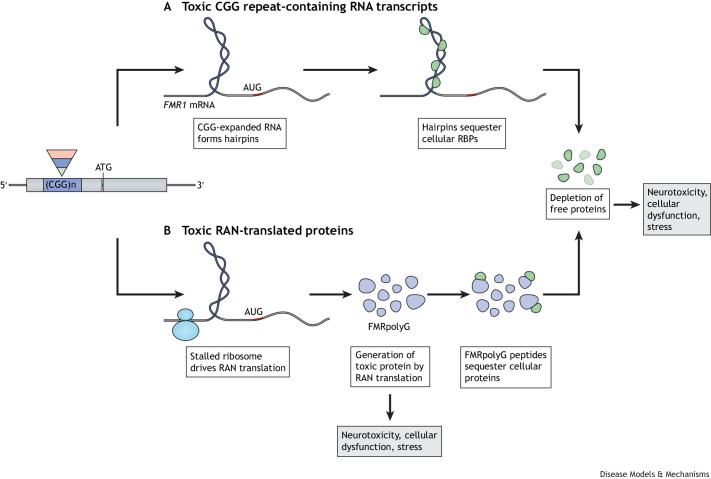
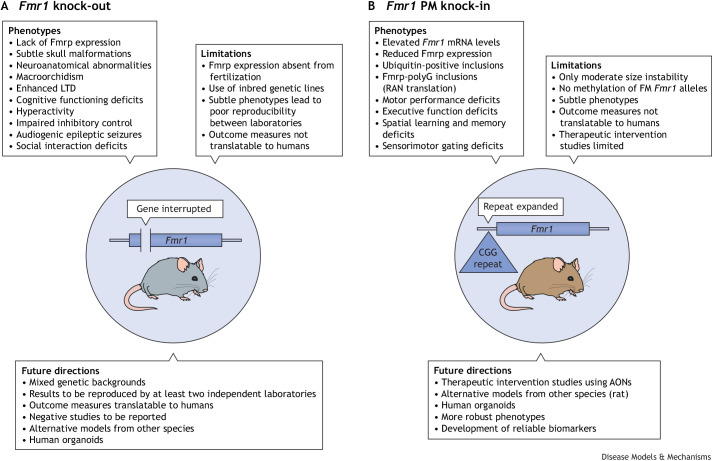
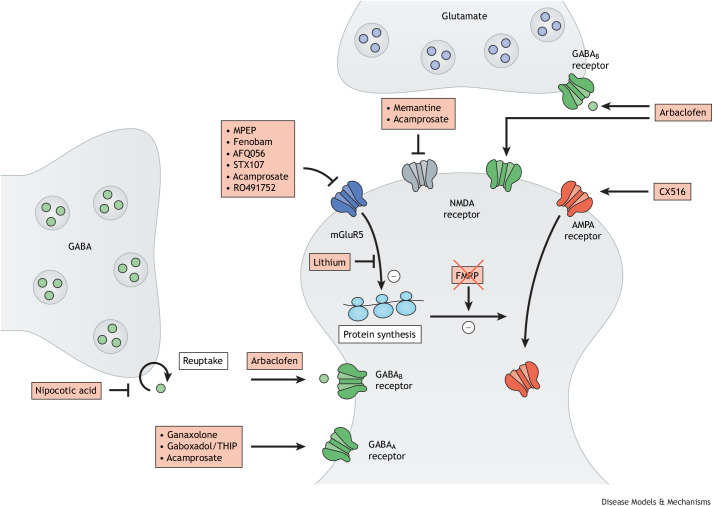
The fragile X-related disorders are an important group of hereditary disorders that are caused by expanded CGG repeats in the 5' untranslated region of the FMR1 gene or by mutations in the coding sequence of this gene. Two categories of pathological CGG repeats are associated with these disorders, full mutation alleles and shorter premutation alleles. Individuals with full mutation alleles develop fragile X syndrome, which causes autism and intellectual disability, whereas those with premutation alleles, which have shorter CGG expansions, can develop fragile X-associated tremor/ataxia syndrome, a progressive neurodegenerative disease. Thus, fragile X-related disorders can manifest as neurodegenerative or neurodevelopmental disorders, depending on the size of the repeat expansion. Here, we review mouse models of fragile X-related disorders and discuss how they have informed our understanding of neurodegenerative and neurodevelopmental disorders. We also assess the translational value of these models for developing rational targeted therapies for intellectual disability and autism disorders.
Keywords: FMR1; Fragile X syndrome; Fragile X-associated tremor/ataxia syndrome; Mouse models.
© 2023. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
References
-
- Allingham-Hawkins, D. J., Babul-Hirji, R., Chitayat, D., Holden, J. J., Yang, K. T., Lee, C., Hudson, R., Gorwill, H., Nolin, S. L., Glicksman, A.et al. (1999). Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study--preliminary data. Am. J. Med. Genet 83, 322-325. 10.1002/(SICI)1096-8628(19990402)83:4<322::AID-AJMG17>3.0.CO;2-B - DOI - PMC - PubMed
-
- Asiminas, A., Jackson, A. D., Louros, S. R., Till, S. M., Spano, T., Dando, O., Bear, M. F., Chattarji, S., Hardingham, G. E., Osterweil, E. K.et al. (2019). Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome. Sci. Transl. Med. 11, eaao0498. 10.1126/scitranslmed.aao0498 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
