

A Homozygous PPP1R21 Splice Variant Associated with Severe Developmental Delay, Absence of Speech, and Muscle Weakness Leads to Activated Proteasome Function
- PMID: 36692708
- PMCID: PMC10039818
- DOI: 10.1007/s12035-023-03219-9
A Homozygous PPP1R21 Splice Variant Associated with Severe Developmental Delay, Absence of Speech, and Muscle Weakness Leads to Activated Proteasome Function
Erratum in
-
Correction: A Homozygous PPP1R21 Splice Variant Associated with Severe Developmental Delay, Absence of Speech, and Muscle Weakness Leads to Activated Proteasome Function.Mol Neurobiol. 2023 Jul;60(7):4164. doi: 10.1007/s12035-023-03319-6. Mol Neurobiol. 2023. PMID: 36941504 Free PMC article. No abstract available.
Abstract
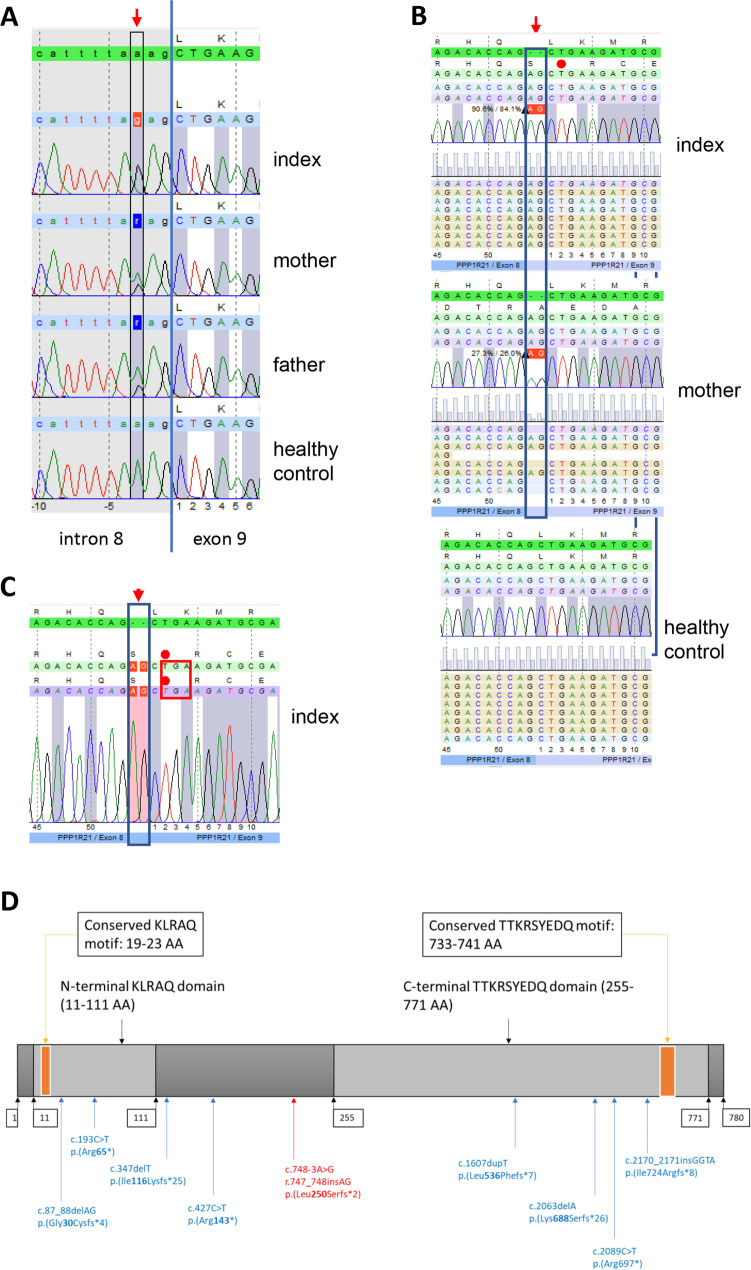
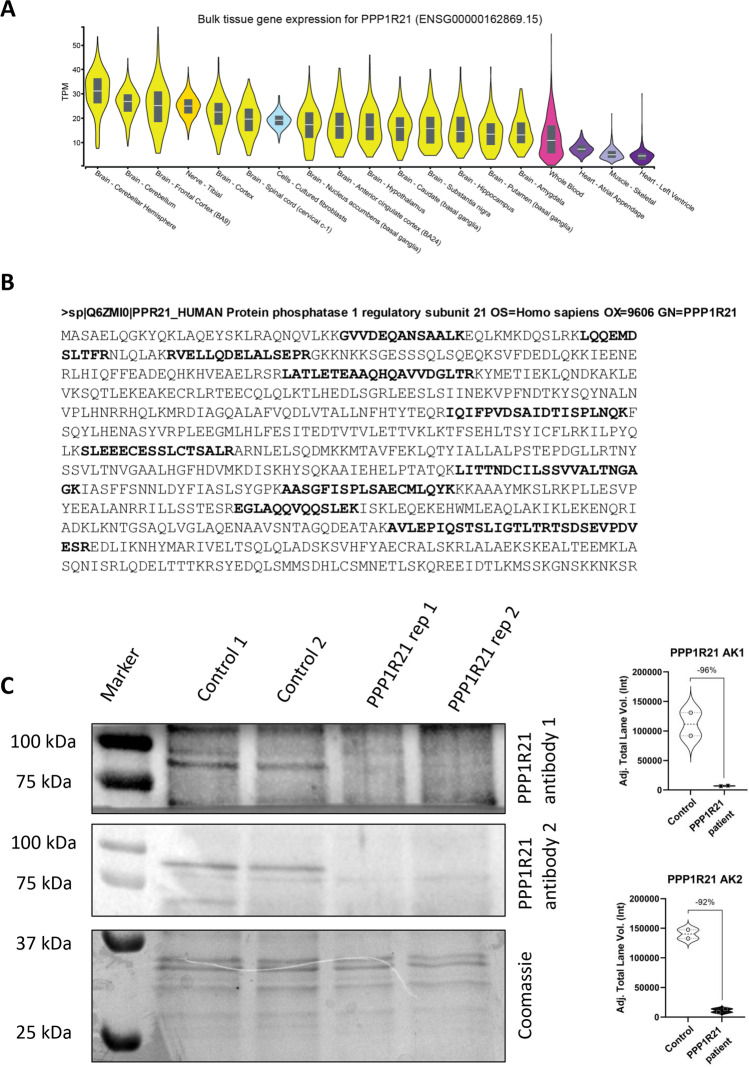
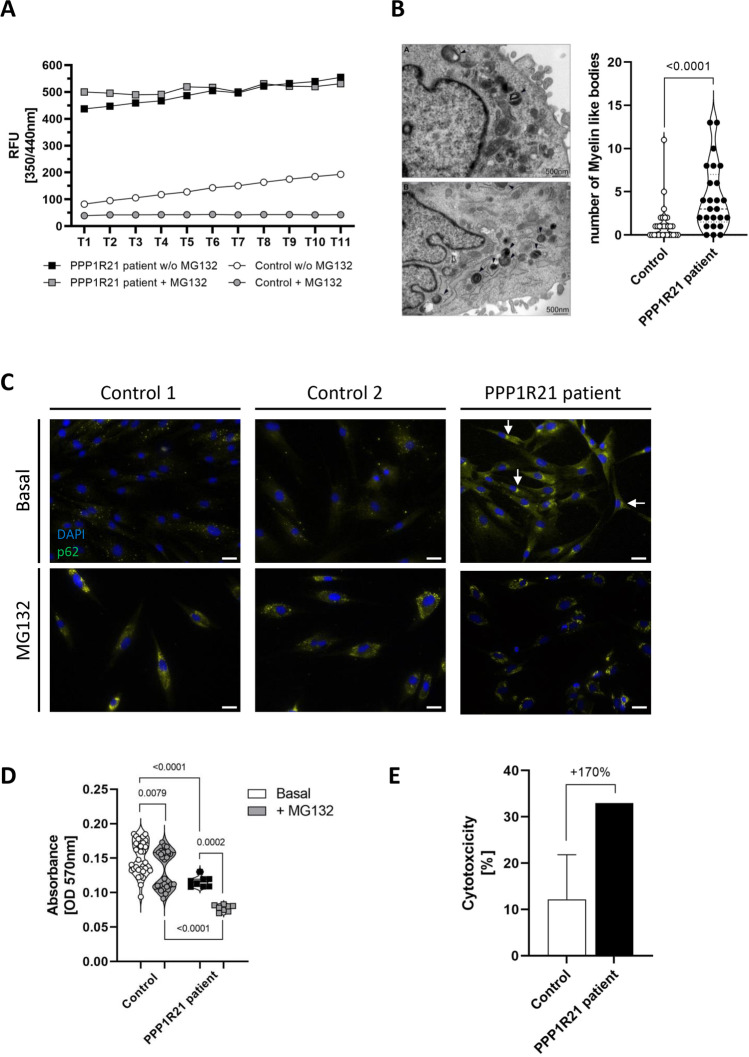
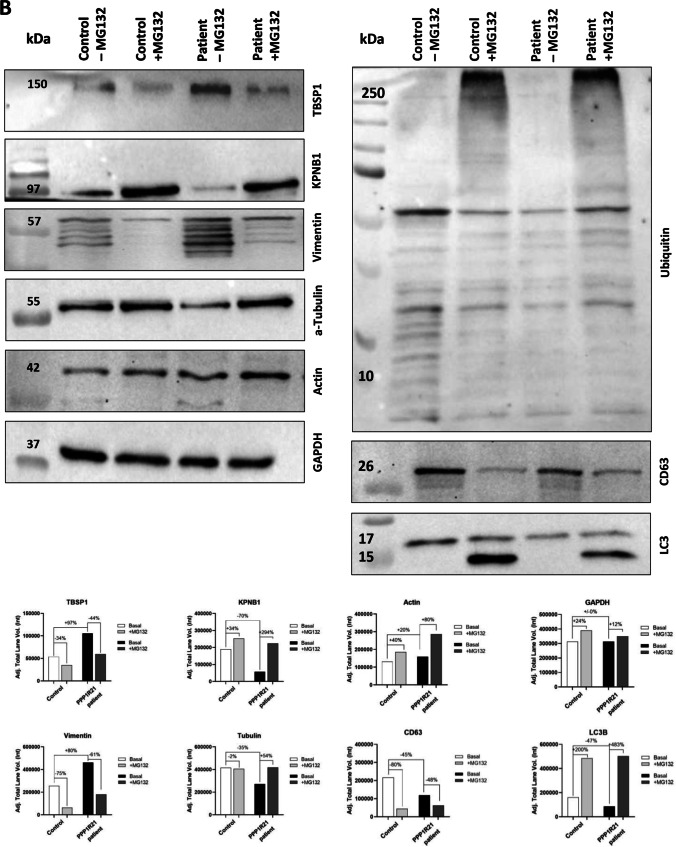
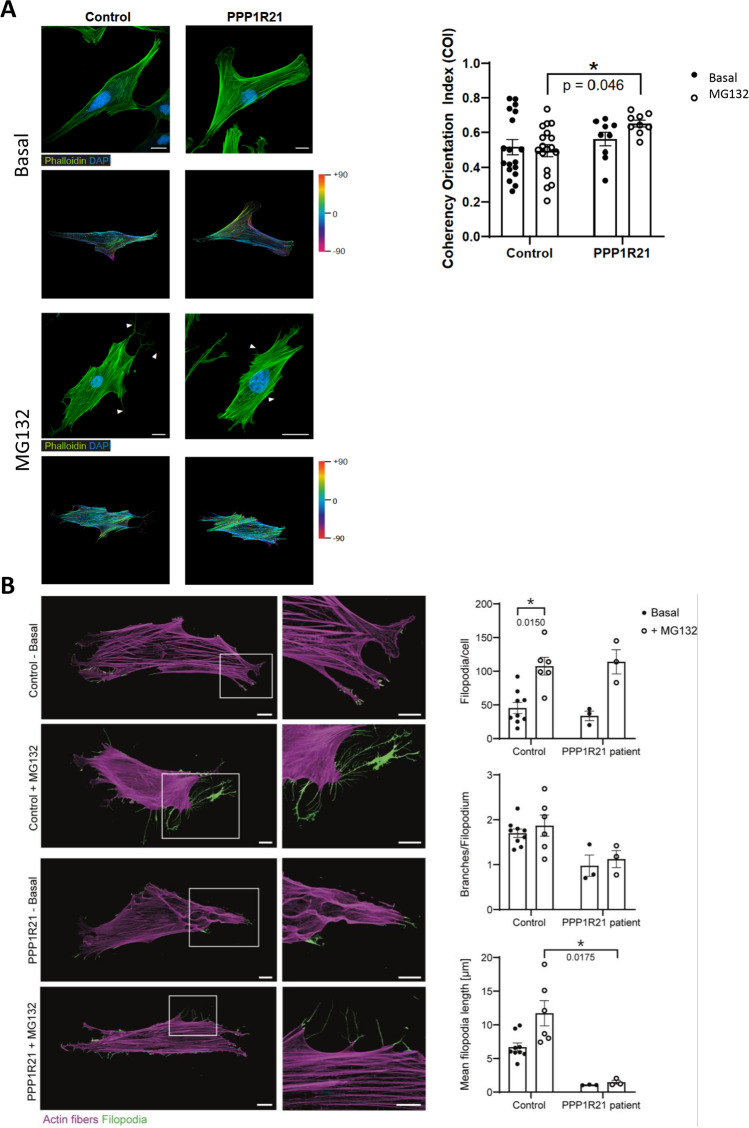
PPP1R21 acts as a co-factor for protein phosphatase 1 (PP1), an important serine/threonine phosphatase known to be essential for cell division, control of glycogen metabolism, protein synthesis, and muscle contractility. Bi-allelic pathogenic variants in PPP1R21 were linked to a neurodevelopmental disorder with hypotonia, facial dysmorphism, and brain abnormalities (NEDHFBA) with pediatric onset. Functional studies unraveled impaired vesicular transport as being part of PPP1R21-related pathomechanism. To decipher further the pathophysiological processes leading to the clinical manifestation of NEDHFBA, we investigated the proteomic signature of fibroblasts derived from the first NEDHFBA patient harboring a splice-site mutation in PPP1R21 and presenting with a milder phenotype. Proteomic findings and further functional studies demonstrate a profound activation of the ubiquitin-proteasome system with presence of protein aggregates and impact on cellular fitness and moreover suggest a cross-link between activation of the proteolytic system and cytoskeletal architecture (including filopodia) as exemplified on paradigmatic proteins including actin, thus extending the pathophysiological spectrum of the disease. In addition, the proteomic signature of PPP1R21-mutant fibroblasts displayed a dysregulation of a variety of proteins of neurological relevance. This includes increase proteins which might act toward antagonization of cellular stress burden in terms of pro-survival, a molecular finding which might accord with the presentation of a milder phenotype of our NEDHFBA patient.
Keywords: Fibroblast cytoskeleton; Fibroblast electron microscopy; Fibroblast filopodia; Fibroblast proteomics; NEDHFBA; PPP1R21; Proteasome.
© 2023. The Author(s).
Conflict of interest statement
Seven of the authors of this publication are members of the European Reference Network for Neuromuscular Diseases-Project ID N° 870177. Three authors of this study are part of the Center for Translational Neuro- and Behavioral Sciences (C-NTBS). The other authors declare no competing interests.
Figures


Similar articles
-
PPP1R21 homozygous null variants associated with developmental delay, muscle weakness, distinctive facial features, and brain abnormalities.Clin Genet. 2018 Oct;94(3-4):351-355. doi: 10.1111/cge.13387. Epub 2018 Jun 25. Clin Genet. 2018. PMID: 29808498
-
PPP1R21-related syndromic intellectual disability: Report of an adult patient and review.Am J Med Genet A. 2020 Dec;182(12):3014-3022. doi: 10.1002/ajmg.a.61889. Epub 2020 Sep 27. Am J Med Genet A. 2020. PMID: 32985083 Review.
-
Correction: A Homozygous PPP1R21 Splice Variant Associated with Severe Developmental Delay, Absence of Speech, and Muscle Weakness Leads to Activated Proteasome Function.Mol Neurobiol. 2023 Jul;60(7):4164. doi: 10.1007/s12035-023-03319-6. Mol Neurobiol. 2023. PMID: 36941504 Free PMC article. No abstract available.
-
Biallelic loss of function variants in PPP1R21 cause a neurodevelopmental syndrome with impaired endocytic function.Hum Mutat. 2019 Mar;40(3):267-280. doi: 10.1002/humu.23694. Epub 2018 Dec 25. Hum Mutat. 2019. PMID: 30520571 Free PMC article.
-
Craniofacial dysmorphism, skeletal anomalies, and impaired intellectual development syndrome-1 in two new patients with the same homozygous TMCO1 variant and review of the literature.Eur J Med Genet. 2023 Mar;66(3):104715. doi: 10.1016/j.ejmg.2023.104715. Epub 2023 Jan 25. Eur J Med Genet. 2023. PMID: 36708876 Review.
Cited by
-
Expanding the phenotype of PPP1R21-related neurodevelopmental disorder.Clin Genet. 2024 Jun;105(6):620-629. doi: 10.1111/cge.14492. Epub 2024 Feb 14. Clin Genet. 2024. PMID: 38356149 Free PMC article.
-
Neurogenetic disorders associated with mutations in the FERRY complex: a novel disease class?Biol Open. 2025 Mar 15;14(3):BIO061808. doi: 10.1242/bio.061808. Epub 2025 Mar 10. Biol Open. 2025. PMID: 40062705 Free PMC article. Review.
-
Search for Ancient Selection Traces in Faverolle Chicken Breed (Gallus gallus domesticus) Based on Runs of Homozygosity Analysis.Animals (Basel). 2025 May 20;15(10):1487. doi: 10.3390/ani15101487. Animals (Basel). 2025. PMID: 40427364 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
