

Genome-Wide Analysis of Structural Variants in Parkinson Disease
- PMID: 36695634
- PMCID: PMC10192042
- DOI: 10.1002/ana.26608
Genome-Wide Analysis of Structural Variants in Parkinson Disease
Abstract
Objective: Identification of genetic risk factors for Parkinson disease (PD) has to date been primarily limited to the study of single nucleotide variants, which only represent a small fraction of the genetic variation in the human genome. Consequently, causal variants for most PD risk are not known. Here we focused on structural variants (SVs), which represent a major source of genetic variation in the human genome. We aimed to discover SVs associated with PD risk by performing the first large-scale characterization of SVs in PD.
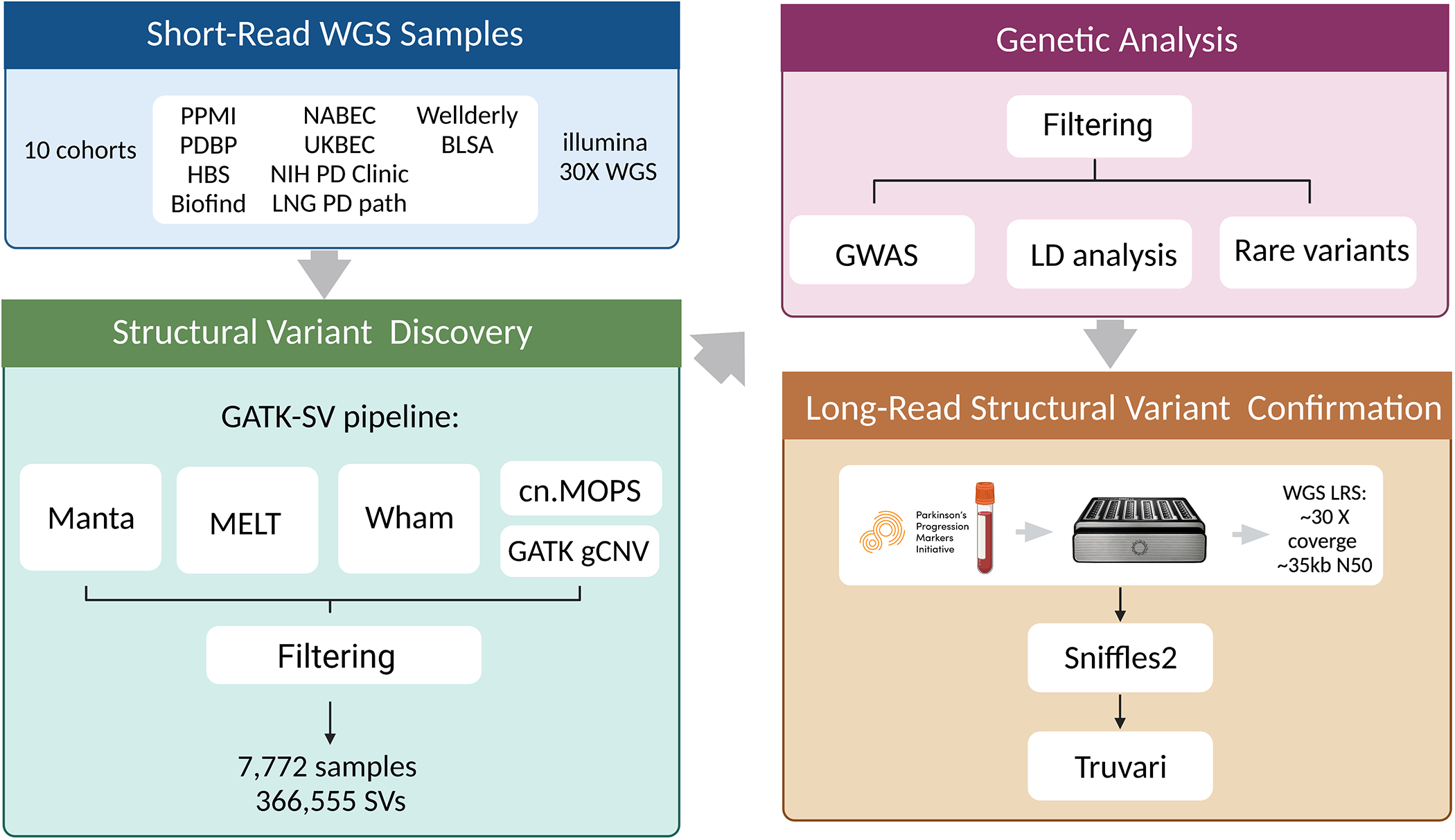
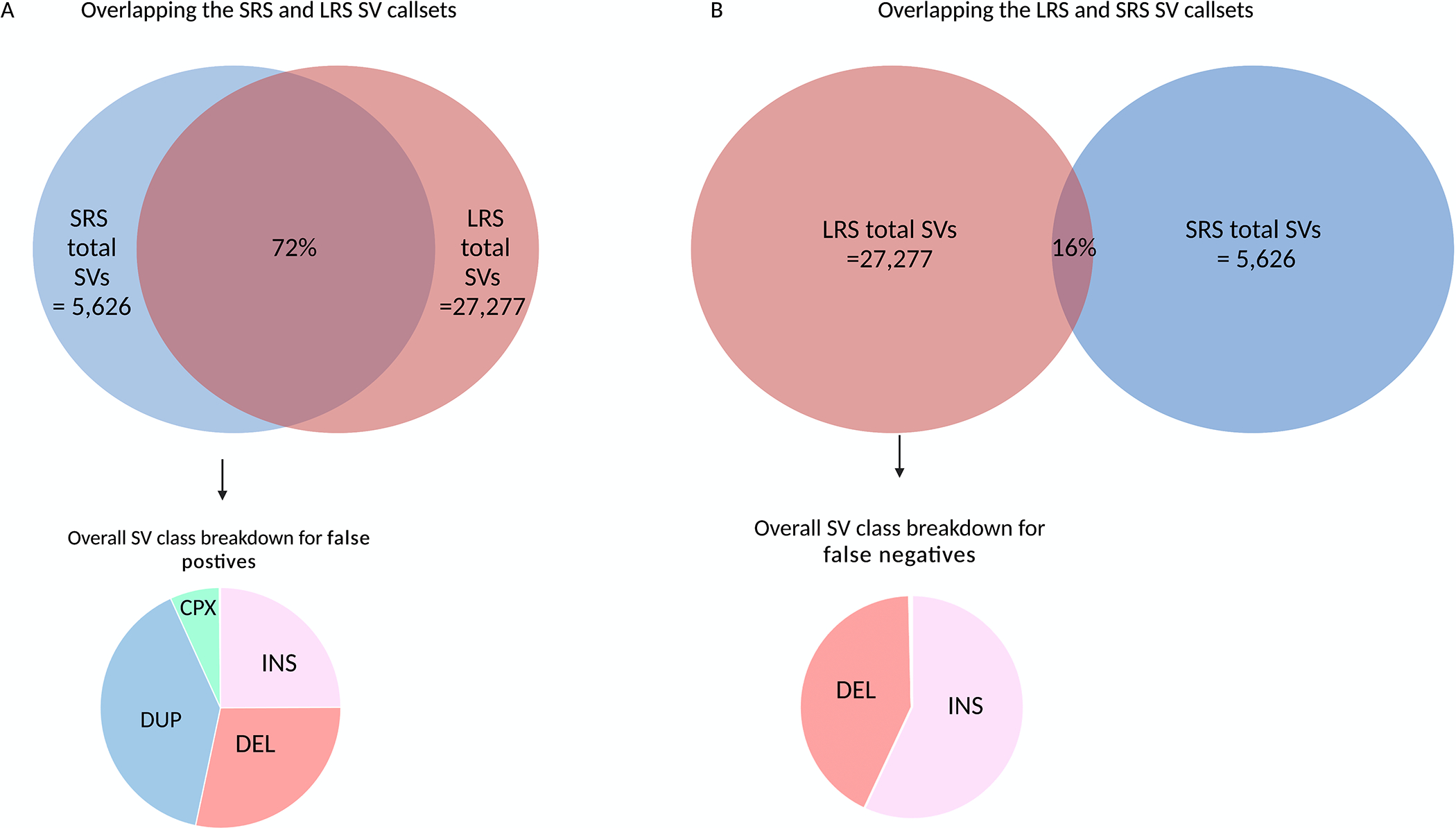
Methods: We leveraged a recently developed computational pipeline to detect and genotype SVs from 7,772 Illumina short-read whole genome sequencing samples. Using this set of SV variants, we performed a genome-wide association study using 2,585 cases and 2,779 controls and identified SVs associated with PD risk. Furthermore, to validate the presence of these variants, we generated a subset of matched whole-genome long-read sequencing data.
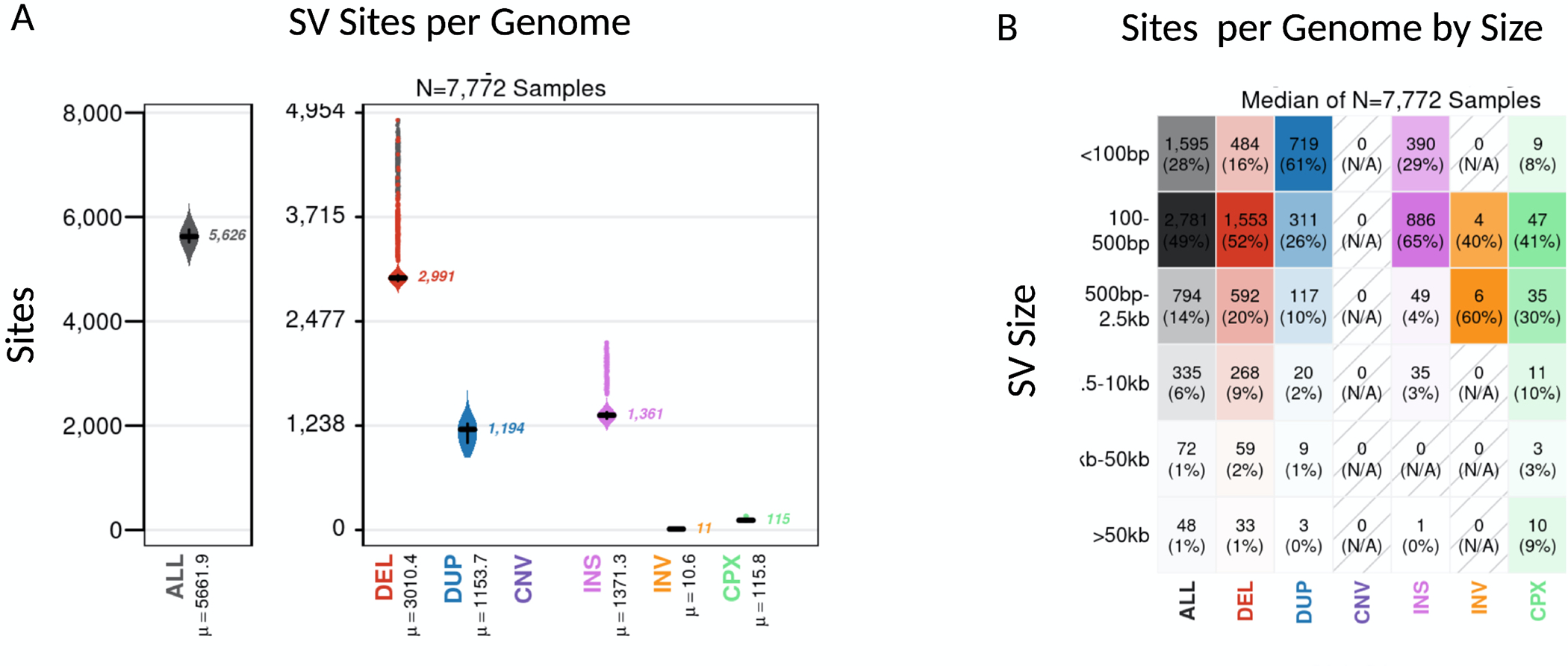
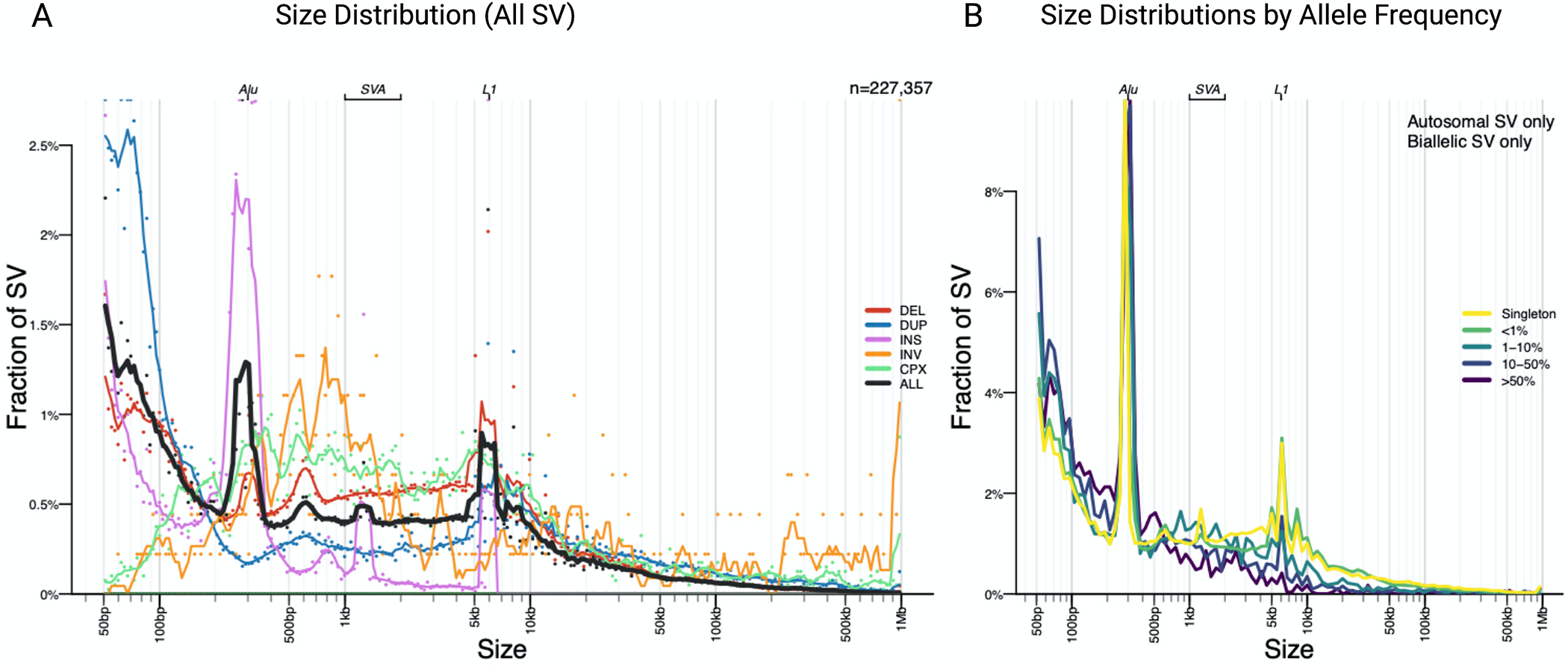
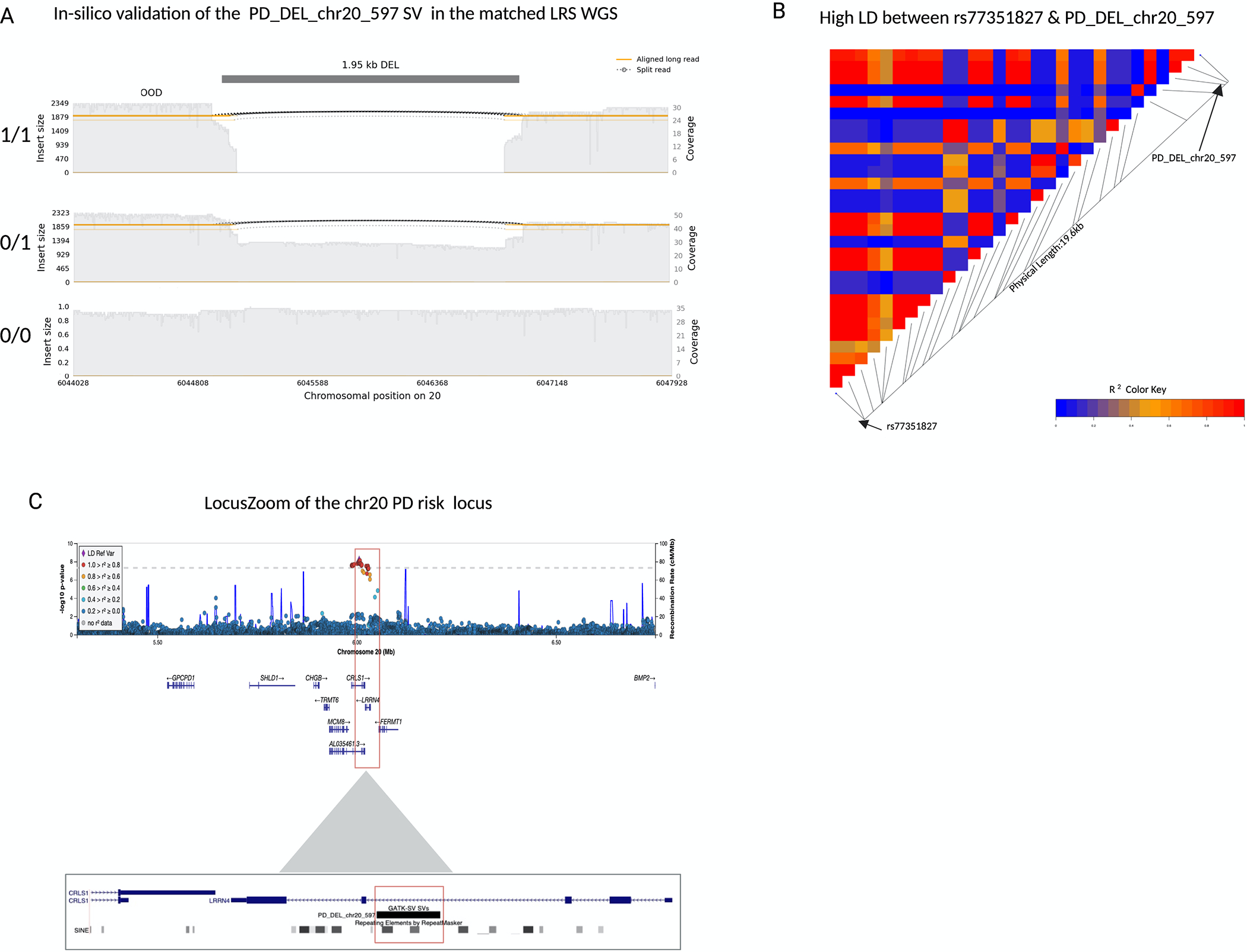
Results: We genotyped and tested 3,154 common SVs, representing over 412 million nucleotides of previously uncatalogued genetic variation. Using long-read sequencing data, we validated the presence of three novel deletion SVs that are associated with risk of PD from our initial association analysis, including a 2 kb intronic deletion within the gene LRRN4.
Interpretation: We identified three SVs associated with genetic risk of PD. This study represents the most comprehensive assessment of the contribution of SVs to the genetic risk of PD to date. ANN NEUROL 2023;93:1012-1022.
© 2023 The Authors. Annals of Neurology published by Wiley Periodicals LLC on behalf of American Neurological Association. This article has been contributed to by U.S. Government employees and their work is in the public domain in the USA.
Conflict of interest statement
Potential Conflicts of Interest
D.V, K.L and M.A.N.’s participation in this project was part of a competitive contract awarded to Data Tecnica International LLC by the National Institutes of Health to support open science research. M.A.N. also currently serves on the scientific advisory board for Clover Therapeutics and is an advisor to Neuron23 Inc. BT currently serves on the Editorial board of Clinical Medicine, JNNP, NBA, is an Associate Editor for Brain and has a collaborative research agreement with Ionis Pharmaceuticals, Roche and Optimeos. FJS receives research support from Illumina, ONT and PacBio. AM works for ONT. M.E.T. receives research funding and/or reagents from Levo Therapeutics, Microsoft Inc., and Illumina Inc.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
