

Dynamic changes in whole genome DNA methylation, chromatin and gene expression during mouse lens differentiation
- PMID: 36698218
- PMCID: PMC9875507
- DOI: 10.1186/s13072-023-00478-7
Dynamic changes in whole genome DNA methylation, chromatin and gene expression during mouse lens differentiation
Abstract
Background: Cellular differentiation is marked by temporally and spatially coordinated gene expression regulated at multiple levels. DNA methylation represents a universal mechanism to control chromatin organization and its accessibility. Cytosine methylation of CpG dinucleotides regulates binding of methylation-sensitive DNA-binding transcription factors within regulatory regions of transcription, including promoters and distal enhancers. Ocular lens differentiation represents an advantageous model system to examine these processes as lens comprises only two cell types, the proliferating lens epithelium and postmitotic lens fiber cells all originating from the epithelium.
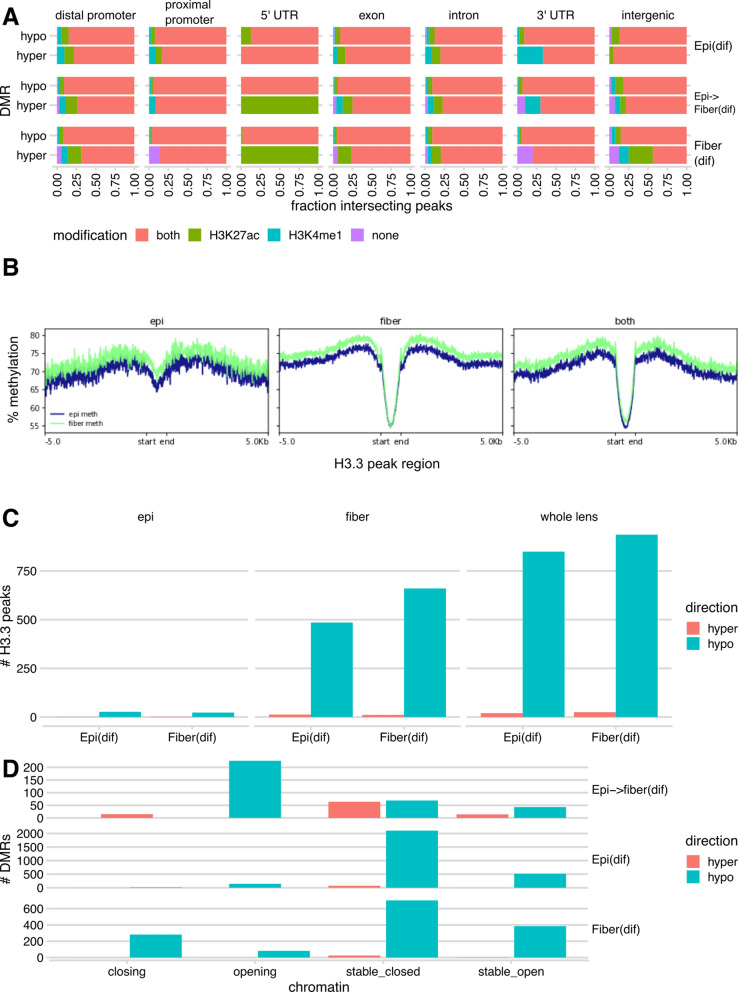
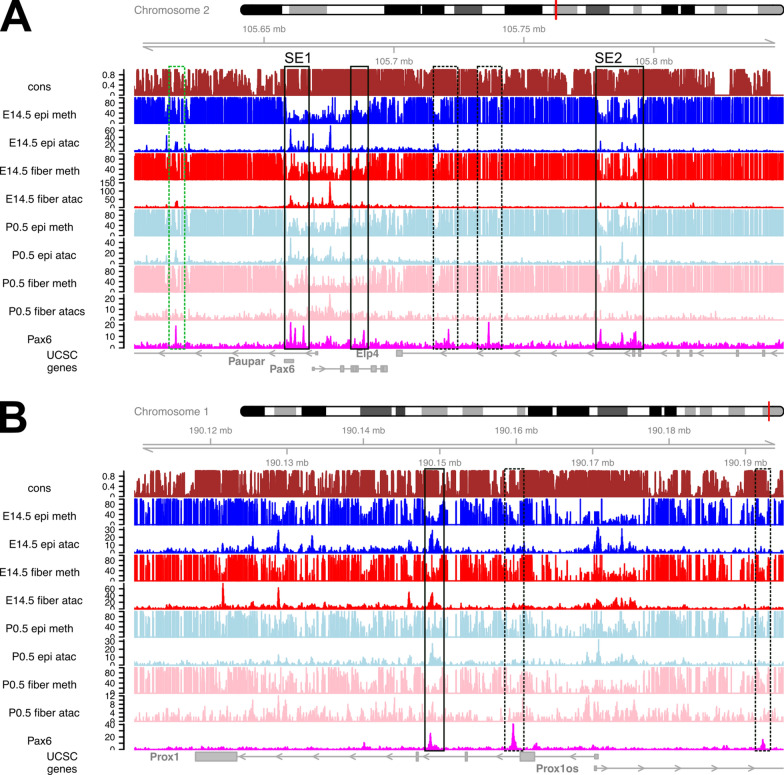
Results: Using whole genome bisulfite sequencing (WGBS) and microdissected lenses, we investigated dynamics of DNA methylation and chromatin changes during mouse lens fiber and epithelium differentiation between embryos (E14.5) and newborns (P0.5). Histone H3.3 variant chromatin landscapes were also generated for both P0.5 lens epithelium and fibers by chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq). Tissue-specific features of DNA methylation patterns are demonstrated via comparative studies with embryonic stem (ES) cells and neural progenitor cells (NPCs) at Nanog, Pou5f1, Sox2, Pax6 and Six3 loci. Comparisons with ATAC-seq and RNA-seq data demonstrate that reduced methylation is associated with increased expression of fiber cell abundant genes, including crystallins, intermediate filament (Bfsp1 and Bfsp2) and gap junction proteins (Gja3 and Gja8), marked by high levels of histone H3.3 within their transcribed regions. Interestingly, Pax6-binding sites exhibited predominantly DNA hypomethylation in lens chromatin. In vitro binding of Pax6 proteins showed Pax6's ability to interact with sites containing one or two methylated CpG dinucleotides.
Conclusions: Our study has generated the first data on methylation changes between two different stages of mammalian lens development and linked these data with chromatin accessibility maps, presence of histone H3.3 and gene expression. Reduced DNA methylation correlates with expression of important genes involved in lens morphogenesis and lens fiber cell differentiation.
Keywords: ATAC-seq; DNA methylation; Differentiation; Gene regulation; Histone H3.3; Lens; Open chromatin; Pax6; RNA-seq.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures












References
-
- Clark SJ, Argelaguet R, Kapourani C-A, Stubbs TM, Lee HJ, Alda-Catalinas C, Krueger F, Sanguinetti G, Kelsey G, Marioni JC, Stegle O, Reik W. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9:781. doi: 10.1038/s41467-018-03149-4. - DOI - PMC - PubMed
-
- Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA, Imaz-Rosshandler I, Lohoff T, Xiang Y, Hanna CW, Smallwood S, Ibarra-Soria X, Buettner F, Sanguinetti G, Xie W, Krueger F, Göttgens B, Rugg-Gunn PJ, Kelsey G, Dean W, Nichols J, Stegle O, Marioni JC, Reik W. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019;2019(576):487–491. doi: 10.1038/s41586-019-1825-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
