

Rise of the planet of rare anemias: An update on emerging treatment strategies
- PMID: 36698833
- PMCID: PMC9868867
- DOI: 10.3389/fmed.2022.1097426
Rise of the planet of rare anemias: An update on emerging treatment strategies
Abstract
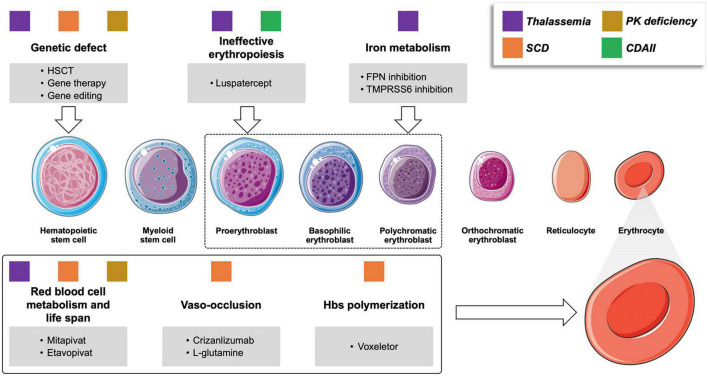
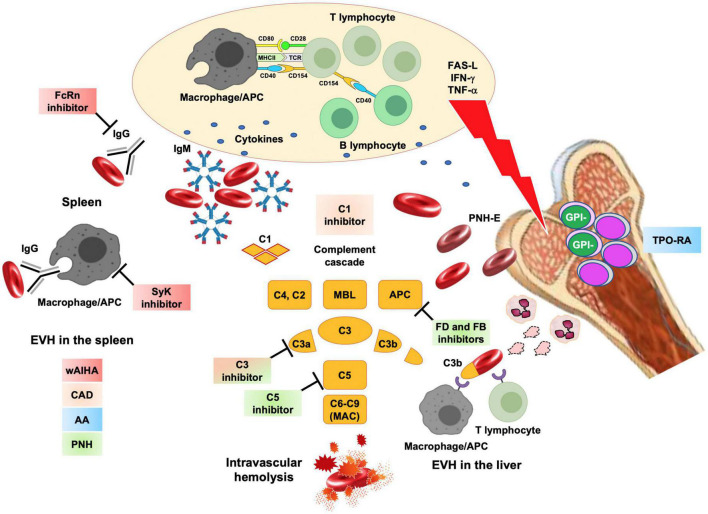
Therapeutic options for rare congenital (hemoglobinopathies, membrane and enzyme defects, congenital dyserythropoietic anemia) and acquired anemias [warm autoimmune hemolytic anemia (wAIHA), cold agglutinin disease CAD, paroxysmal nocturnal hemoglobinuria (PNH), and aplastic anemia (AA)] are rapidly expanding. The use of luspatercept, mitapivat and etavopivat in beta-thalassemia and pyruvate kinase deficiency (PKD) improves transfusion dependence, alleviating iron overload and long-term complications. Voxelotor, mitapivat, and etavopivat reduce vaso-occlusive crises in sickle cell disease (SCD). Gene therapy represents a fascinating approach, although patient selection, the toxicity of the conditioning regimens, and the possible long-term safety are still open issues. For acquired forms, wAIHA and CAD will soon benefit from targeted therapies beyond rituximab, including B-cell/plasma cell targeting agents (parsaclisib, rilzabrutinib, and isatuximab for wAIHA), complement inhibitors (pegcetacoplan and sutimlimab for CAD, ANX005 for wAIHA with complement activation), and inhibitors of extravascular hemolysis in the reticuloendothelial system (fostamatinib and FcRn inhibitors in wAIHA). PNH treatment is moving from the intravenous anti-C5 eculizumab to its long-term analog ravulizumab, and to subcutaneous and oral proximal inhibitors (anti-C3 pegcetacoplan, factor D and factor B inhibitors danicopan and iptacopan). These drugs have the potential to improve patient convenience and ameliorate residual anemia, although patient compliance becomes pivotal, and long-term safety requires further investigation. Finally, the addition of eltrombopag significantly ameliorated AA outcomes, and data regarding the alternative agent romiplostim are emerging. The accelerated evolution of treatment strategies will need further effort to identify the best candidate for each treatment in the precision medicine era.
Keywords: aplastic anemia; autoimmune hemolytic anemia; beta-thalassemia; cold agglutinin disease; congenital hemolytic anemias; paroxysmal nocturnal hemoglobinuria; pyruvate kinase deficiency; sickle cell disease.
Copyright © 2023 Fattizzo and Motta.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous