

Cardiac repair after myocardial infarction: A two-sided role of inflammation-mediated
- PMID: 36698953
- PMCID: PMC9868426
- DOI: 10.3389/fcvm.2022.1077290
Cardiac repair after myocardial infarction: A two-sided role of inflammation-mediated
Abstract
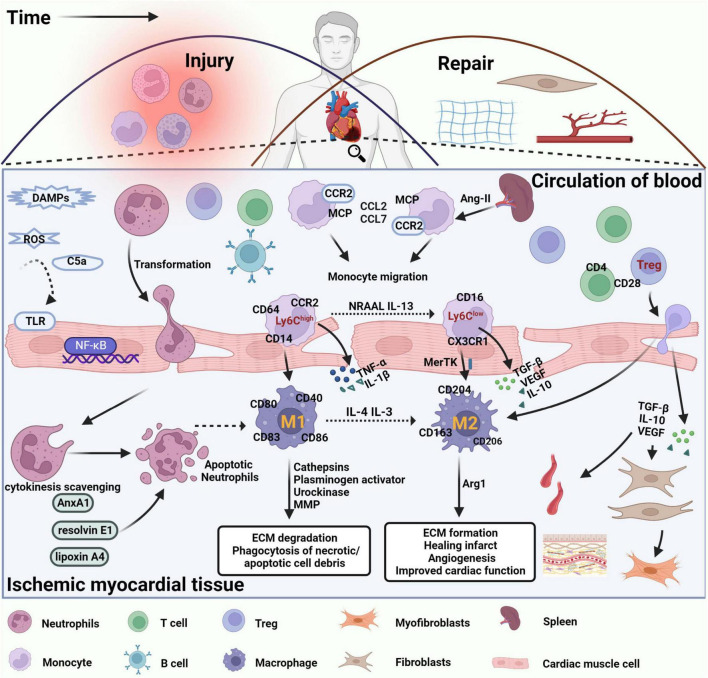
Myocardial infarction is the leading cause of death and disability worldwide, and the development of new treatments can help reduce the size of myocardial infarction and prevent adverse cardiovascular events. Cardiac repair after myocardial infarction can effectively remove necrotic tissue, induce neovascularization, and ultimately replace granulation tissue. Cardiac inflammation is the primary determinant of whether beneficial cardiac repair occurs after myocardial infarction. Immune cells mediate inflammatory responses and play a dual role in injury and protection during cardiac repair. After myocardial infarction, genetic ablation or blocking of anti-inflammatory pathways is often harmful. However, enhancing endogenous anti-inflammatory pathways or blocking endogenous pro-inflammatory pathways may improve cardiac repair after myocardial infarction. A deficiency of neutrophils or monocytes does not improve overall cardiac function after myocardial infarction but worsens it and aggravates cardiac fibrosis. Several factors are critical in regulating inflammatory genes and immune cells' phenotypes, including DNA methylation, histone modifications, and non-coding RNAs. Therefore, strict control and timely suppression of the inflammatory response, finding a balance between inflammatory cells, preventing excessive tissue degradation, and avoiding infarct expansion can effectively reduce the occurrence of adverse cardiovascular events after myocardial infarction. This article reviews the involvement of neutrophils, monocytes, macrophages, and regulatory T cells in cardiac repair after myocardial infarction. After myocardial infarction, neutrophils are the first to be recruited to the damaged site to engulf necrotic cell debris and secrete chemokines that enhance monocyte recruitment. Monocytes then infiltrate the infarct site and differentiate into macrophages and they release proteases and cytokines that are harmful to surviving myocardial cells in the pre-infarct period. As time progresses, apoptotic neutrophils are cleared, the recruitment of anti-inflammatory monocyte subsets, the polarization of macrophages toward the repair phenotype, and infiltration of regulatory T cells, which secrete anti-inflammatory factors that stimulate angiogenesis and granulation tissue formation for cardiac repair. We also explored how epigenetic modifications regulate the phenotype of inflammatory genes and immune cells to promote cardiac repair after myocardial infarction. This paper also elucidates the roles of alarmin S100A8/A9, secreted frizzled-related protein 1, and podoplanin in the inflammatory response and cardiac repair after myocardial infarction.
Keywords: cardiac repair; epigenetic modifications; inflammation; inflammatory cells; myocardial infarction; two-way adjustment.
Copyright © 2023 Li, Yan, Fan, Fan, Li, Qi and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
The immune system and cardiac repair.Pharmacol Res. 2008 Aug;58(2):88-111. doi: 10.1016/j.phrs.2008.06.007. Epub 2008 Jun 24. Pharmacol Res. 2008. PMID: 18620057 Free PMC article. Review.
-
Harnessing the early post-injury inflammatory responses for cardiac regeneration.J Biomed Sci. 2017 Jan 13;24(1):7. doi: 10.1186/s12929-017-0315-2. J Biomed Sci. 2017. PMID: 28086885 Free PMC article. Review.
-
The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis.Circ Res. 2016 Jun 24;119(1):91-112. doi: 10.1161/CIRCRESAHA.116.303577. Circ Res. 2016. PMID: 27340270 Free PMC article. Review.
-
The inflammatory response and cardiac repair after myocardial infarction.Korean Circ J. 2009 Oct;39(10):393-8. doi: 10.4070/kcj.2009.39.10.393. Epub 2009 Oct 28. Korean Circ J. 2009. PMID: 19949583 Free PMC article.
-
Anti-inflammatory mechanisms and therapeutic opportunities in myocardial infarct healing.J Mol Med (Berl). 2012 Apr;90(4):361-9. doi: 10.1007/s00109-011-0847-y. Epub 2012 Jan 7. J Mol Med (Berl). 2012. PMID: 22228177 Review.
Cited by
-
Comparison of the proteomic landscape in experimental ischemia reperfusion with versus without ischemic preconditioning.Sci Rep. 2025 Apr 7;15(1):11836. doi: 10.1038/s41598-025-90735-4. Sci Rep. 2025. PMID: 40195349 Free PMC article.
-
Identification of key proteins and pathways in myocardial infarction using machine learning approaches.Sci Rep. 2025 Jun 4;15(1):19530. doi: 10.1038/s41598-025-04401-w. Sci Rep. 2025. PMID: 40467768 Free PMC article.
-
Targeted Treatment of Myocardial Infarction by Macrophage Membrane Coated with Resveratrol Nanoparticles.ACS Omega. 2024 Nov 11;9(47):47145-47155. doi: 10.1021/acsomega.4c07573. eCollection 2024 Nov 26. ACS Omega. 2024. PMID: 39619553 Free PMC article.
-
Endocrine and metabolic alterations in response to systemic inflammation and sepsis: a review article.Mol Med. 2025 Jan 21;31(1):16. doi: 10.1186/s10020-025-01074-z. Mol Med. 2025. PMID: 39838305 Free PMC article. Review.
-
Extracellular vesicle therapeutics for cardiac repair.J Mol Cell Cardiol. 2025 Feb;199:12-32. doi: 10.1016/j.yjmcc.2024.11.005. Epub 2024 Nov 26. J Mol Cell Cardiol. 2025. PMID: 39603560 Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials