

Modeling nonsegmented negative-strand RNA virus (NNSV) transcription with ejective polymerase collisions and biased diffusion
- PMID: 36699700
- PMCID: PMC9868645
- DOI: 10.3389/fmolb.2022.1095193
Modeling nonsegmented negative-strand RNA virus (NNSV) transcription with ejective polymerase collisions and biased diffusion
Abstract
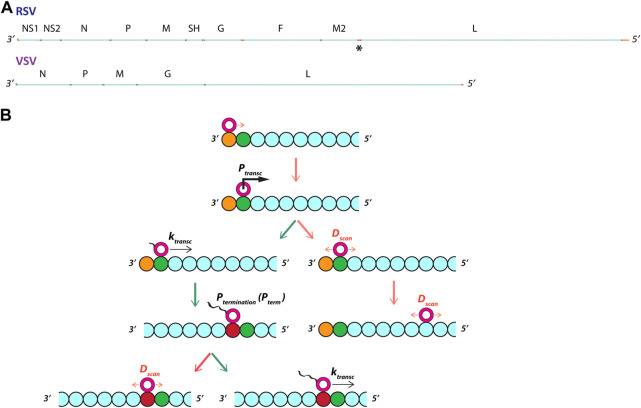
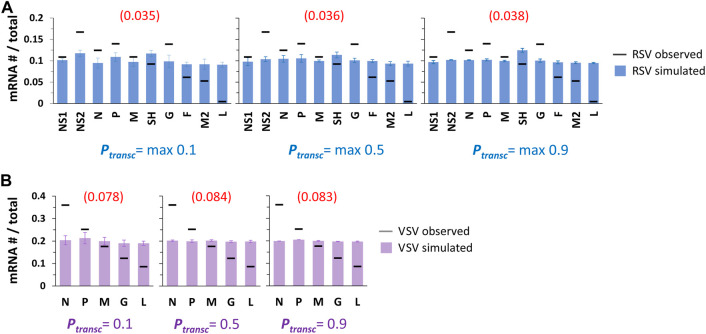
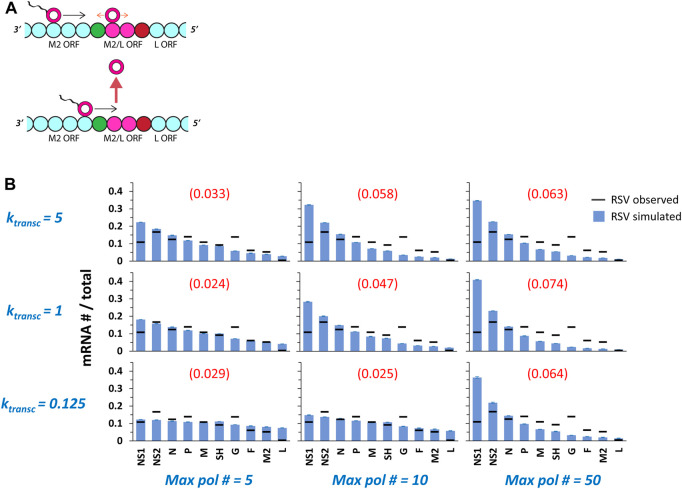
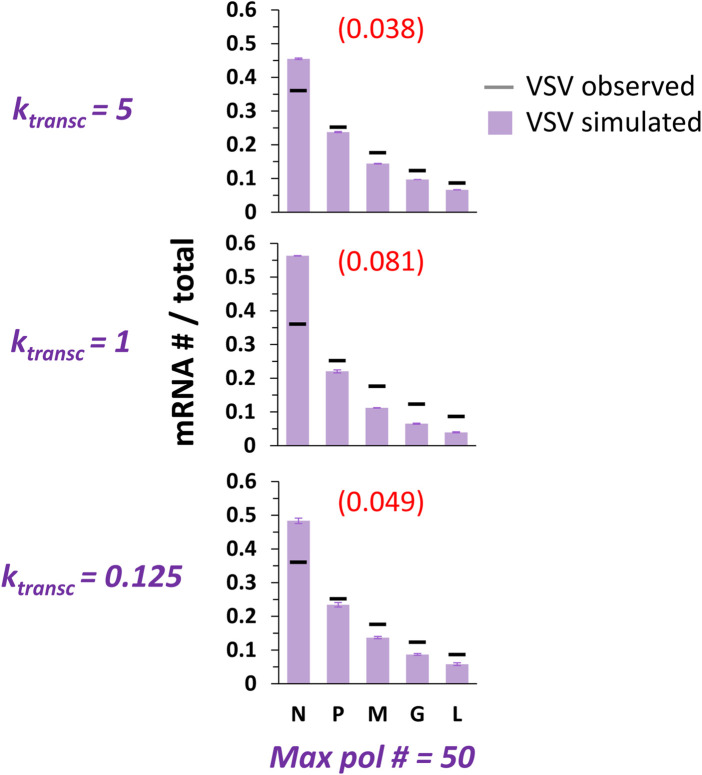
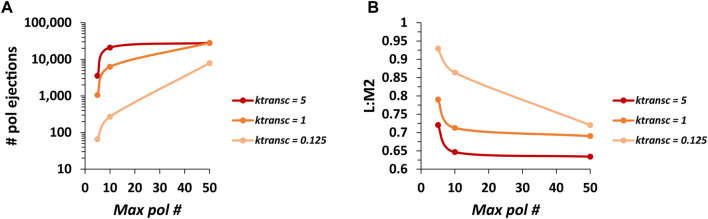
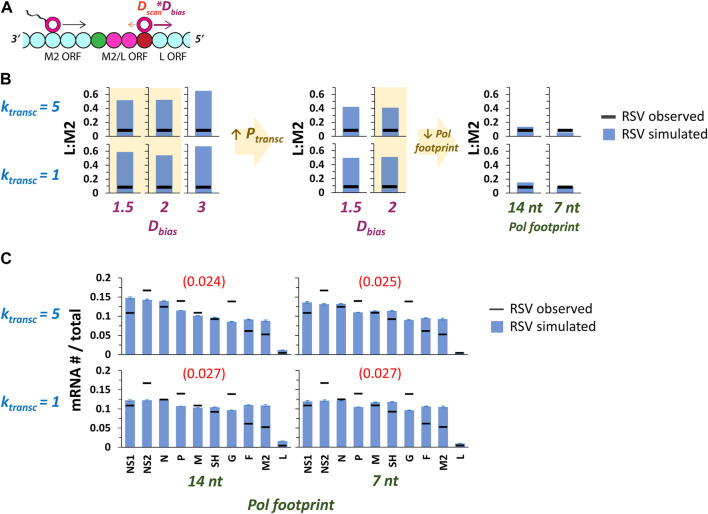
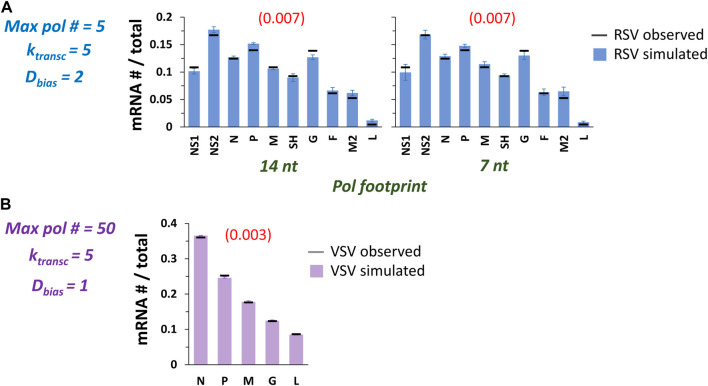
Infections by non-segmented negative-strand RNA viruses (NNSV) are widely thought to entail gradient gene expression from the well-established existence of a single promoter at the 3' end of the viral genome and the assumption of constant transcriptional attenuation between genes. But multiple recent studies show viral mRNA levels in infections by respiratory syncytial virus (RSV), a major human pathogen and member of NNSV, that are inconsistent with a simple gradient. Here we integrate known and newly predicted phenomena into a biophysically reasonable model of NNSV transcription. Our model succeeds in capturing published observations of respiratory syncytial virus and vesicular stomatitis virus (VSV) mRNA levels. We therefore propose a novel understanding of NNSV transcription based on the possibility of ejective polymerase-polymerase collisions and, in the case of RSV, biased polymerase diffusion.
Keywords: RNA viral genome; biased diffusion; polymerase collisions; transcriptional regulation; viral gene expression.
Copyright © 2023 Piedra, Henke, Rajan, Muzny, Doddapaneni, Menon, Hoffman, Ross, Javornik Cregeen, Metcalf, Gibbs, Petrosino, Avadhanula and Piedra.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Aljabr W., Touzelet O., Pollakis G., Wu W., Munday D. C., Hughes M., et al. (2016). Investigating the influence of ribavirin on human respiratory syncytial virus RNA synthesis by using a high-resolution transcriptome sequencing approach. J. Virol. 90 (10), 4876–4888. 10.1128/JVI.02349-15 - DOI - PMC - PubMed
-
- Barr J. N., Tang X., Hinzman E., Shen R., Wertz G. W. (2008). The VSV polymerase can initiate at mRNA start sites located either up or downstream of a transcription termination signal but size of the intervening intergenic region affects efficiency of initiation. Virology 374 (2), 361–370. 10.1016/j.virol.2007.12.023 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources