

Cross center single-cell RNA sequencing study of the immune microenvironment in rapid progressing multiple myeloma
- PMID: 36702834
- PMCID: PMC9879959
- DOI: 10.1038/s41525-022-00340-x
Cross center single-cell RNA sequencing study of the immune microenvironment in rapid progressing multiple myeloma
Abstract
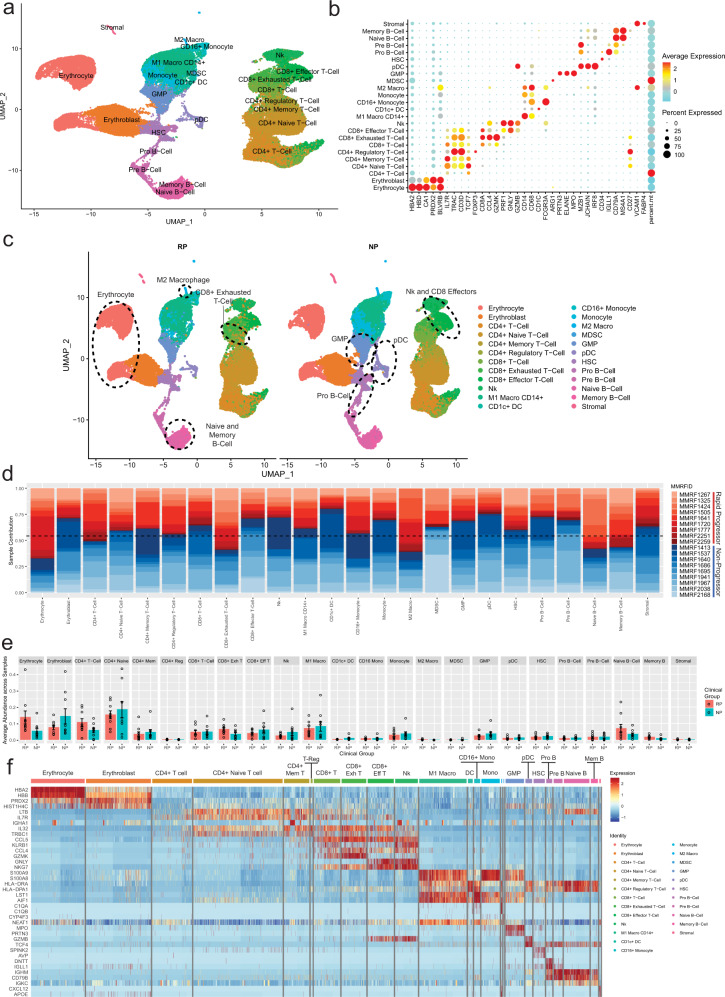
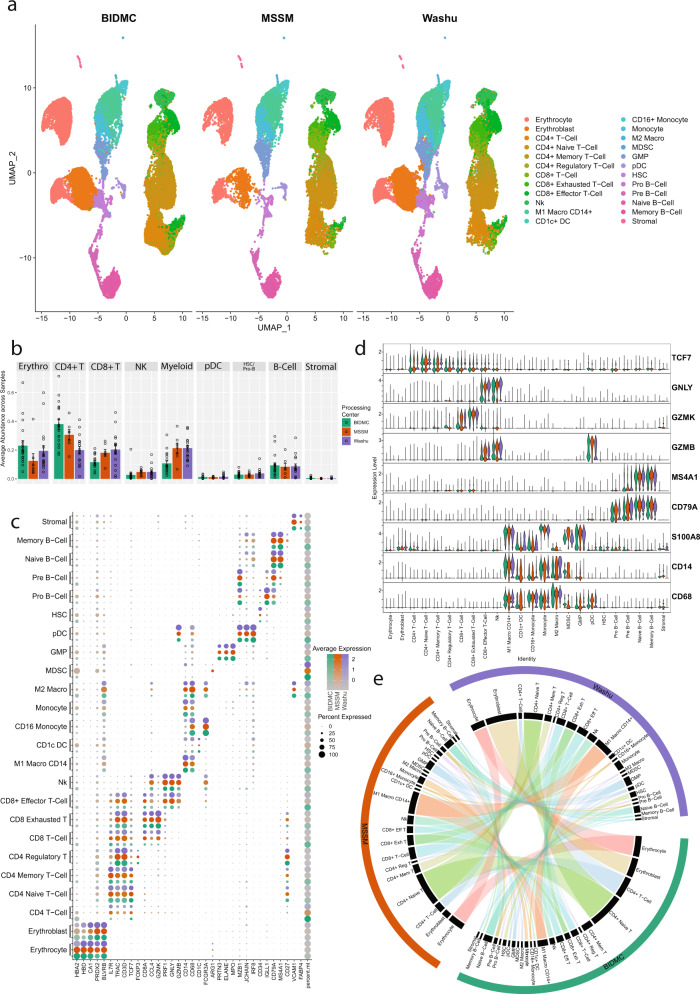
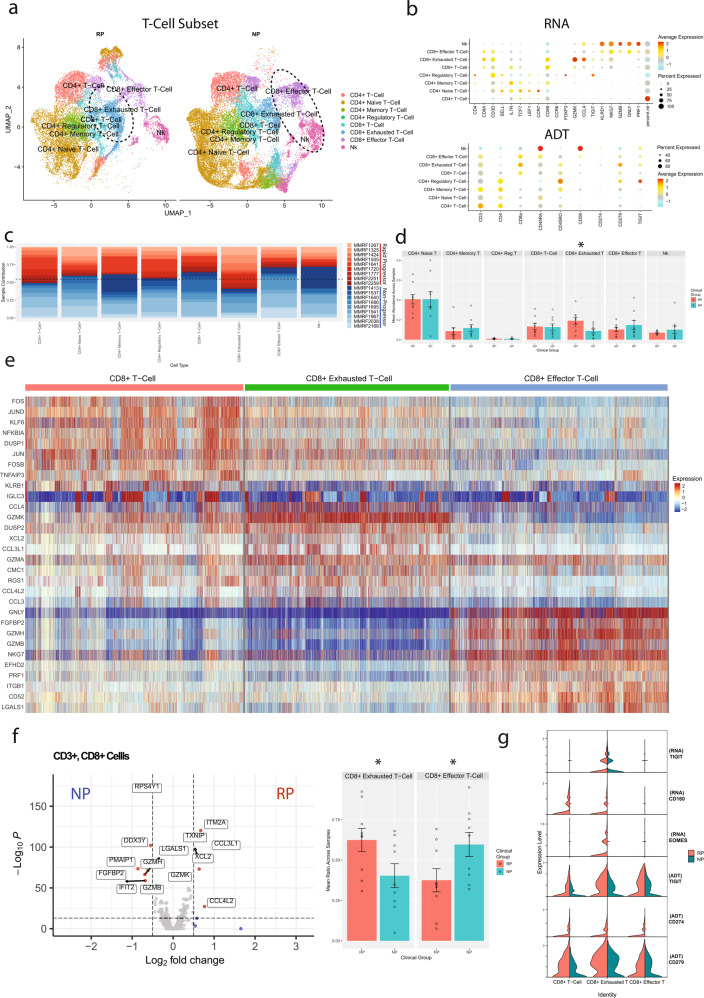
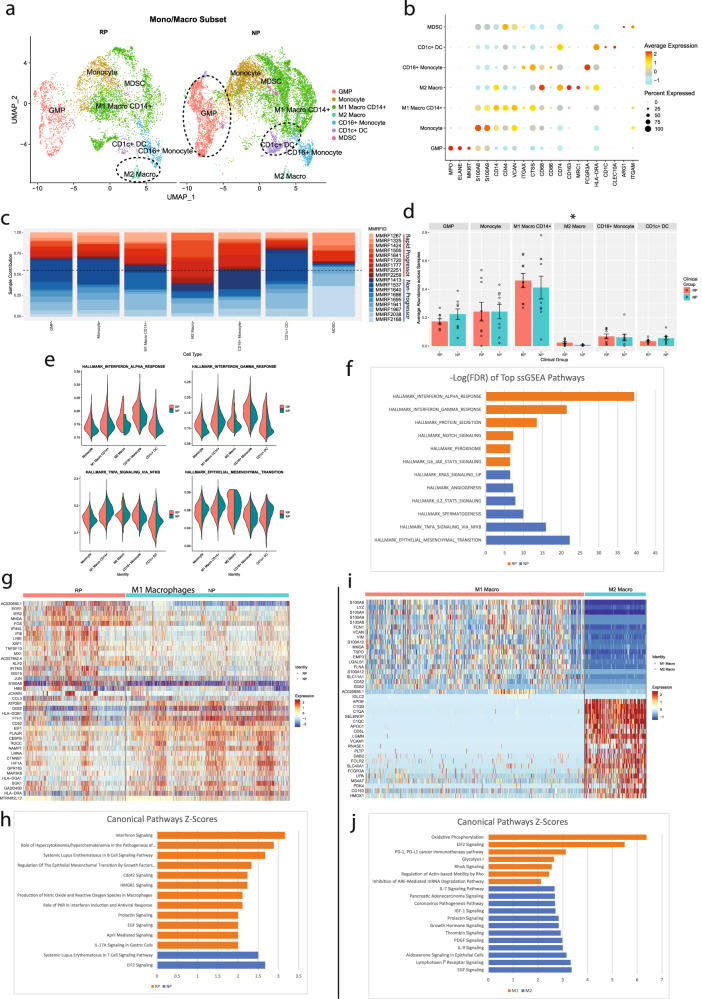
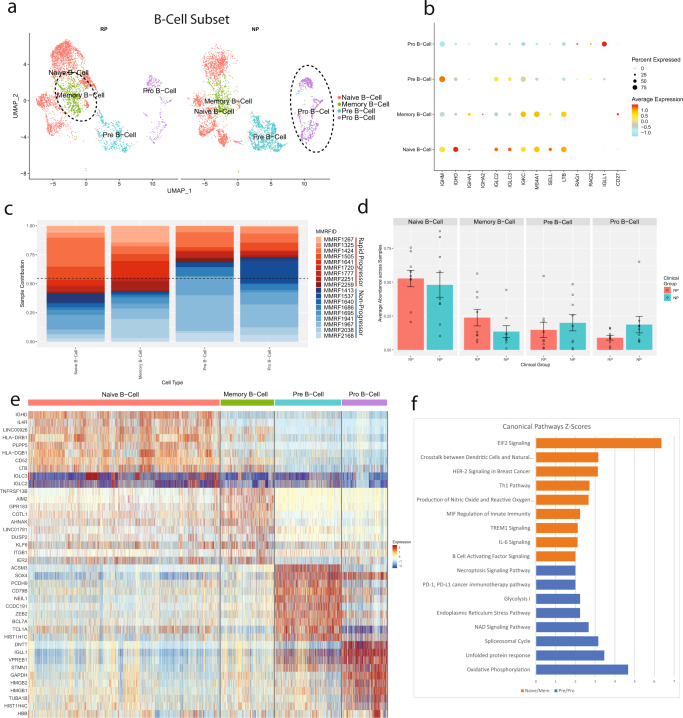
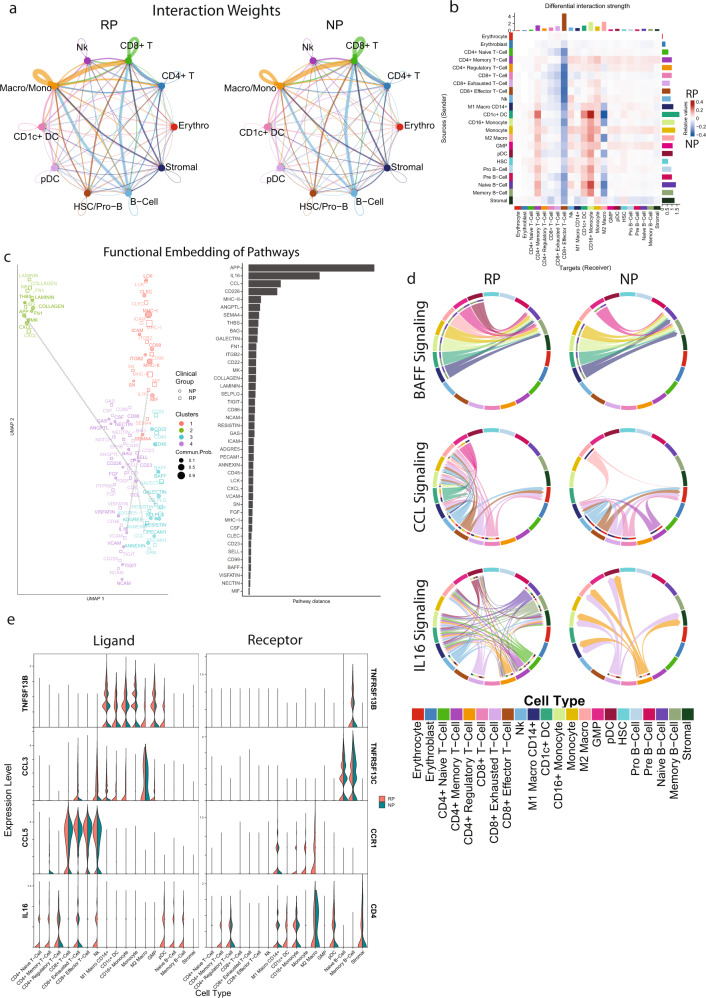
Despite advancements in understanding the pathophysiology of Multiple Myeloma (MM), the cause of rapid progressing disease in a subset of patients is still unclear. MM's progression is facilitated by complex interactions with the surrounding bone marrow (BM) cells, forming a microenvironment that supports tumor growth and drug resistance. Understanding the immune microenvironment is key to identifying factors that promote rapid progression of MM. To accomplish this, we performed a multi-center single-cell RNA sequencing (scRNA-seq) study on 102,207 cells from 48 CD138- BM samples collected at the time of disease diagnosis from 18 patients with either rapid progressing (progression-free survival (PFS) < 18 months) or non-progressing (PFS > 4 years) disease. Comparative analysis of data from three centers demonstrated similar transcriptome profiles and cell type distributions, indicating subtle technical variation in scRNA-seq, opening avenues for an expanded multicenter trial. Rapid progressors depicted significantly higher enrichment of GZMK+ and TIGIT+ exhausted CD8+ T-cells (P = 0.022) along with decreased expression of cytolytic markers (PRF1, GZMB, GNLY). We also observed a significantly higher enrichment of M2 tolerogenic macrophages in rapid progressors and activation of pro-proliferative signaling pathways, such as BAFF, CCL, and IL16. On the other hand, non-progressive patients depicted higher enrichment for immature B Cells (i.e., Pre/Pro B cells), with elevated expression for markers of B cell development (IGLL1, SOX4, DNTT). This multi-center study identifies the enrichment of various pro-tumorigenic cell populations and pathways in those with rapid progressing disease and further validates the robustness of scRNA-seq data generated at different study centers.
© 2023. The Author(s).
Conflict of interest statement
This study was supported by the Multiple Myeloma Research Foundation (MMRF). M.H., S.M., H.J.C., and D.A. are employed by the MMRF, but they did not have a role in the interpretation of the results. S.G. is additionally supported by grants U24 CA224319 and U01 DK124165. The Mount Sinai Human Immune Monitoring Center is supported in part by Cancer Center grant P01 CA196521. S.G. reports consultancy and/or advisory roles for Merck, and OncoMed and research funding from Bristol-Myers Squibb, Genentech, Janssen R&D, Pfizer, Celgene, Takeda, and Regeneron. Other authors declare no competing interests.
Figures
Similar articles
-
Comprehensive Characterization of the Multiple Myeloma Immune Microenvironment Using Integrated scRNA-seq, CyTOF, and CITE-seq Analysis.Cancer Res Commun. 2022 Oct 25;2(10):1255-1265. doi: 10.1158/2767-9764.CRC-22-0022. eCollection 2022 Oct. Cancer Res Commun. 2022. PMID: 36969740 Free PMC article.
-
Identification of potential resistance mechanisms and therapeutic targets for the relapse of BCMA CAR-T therapy in relapsed/refractory multiple myeloma through single-cell sequencing.Exp Hematol Oncol. 2023 May 8;12(1):44. doi: 10.1186/s40164-023-00402-5. Exp Hematol Oncol. 2023. PMID: 37158921 Free PMC article.
-
Characteristics and functions of an atypical inflammation-associated GZMK+GZMB+CD8+ T subset in people living with HIV-1.Mol Immunol. 2024 Sep;173:40-52. doi: 10.1016/j.molimm.2024.07.003. Epub 2024 Jul 24. Mol Immunol. 2024. PMID: 39053388
-
Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy.Cancer Commun (Lond). 2020 Aug;40(8):329-344. doi: 10.1002/cac2.12078. Epub 2020 Jul 12. Cancer Commun (Lond). 2020. PMID: 32654419 Free PMC article. Review.
-
Essential procedures of single-cell RNA sequencing in multiple myeloma and its translational value.Blood Sci. 2023 Nov 2;5(4):221-236. doi: 10.1097/BS9.0000000000000172. eCollection 2023 Oct. Blood Sci. 2023. PMID: 37941914 Free PMC article. Review.
Cited by
-
The Simple prEservatioN of Single cElls method for cryopreservation enables the generation of single-cell immune profiles from whole blood.Front Immunol. 2023 Nov 28;14:1271800. doi: 10.3389/fimmu.2023.1271800. eCollection 2023. Front Immunol. 2023. PMID: 38090590 Free PMC article.
-
CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies.Front Immunol. 2023 Feb 20;14:1101495. doi: 10.3389/fimmu.2023.1101495. eCollection 2023. Front Immunol. 2023. PMID: 36891310 Free PMC article. Review.
-
Integrative single-cell and spatial transcriptome analysis reveals heterogeneity of human liver progenitor cells.Hepatol Commun. 2025 Feb 26;9(3):e0662. doi: 10.1097/HC9.0000000000000662. eCollection 2025 Mar 1. Hepatol Commun. 2025. PMID: 40008906 Free PMC article.
-
A single-cell transcriptomic map of the murine and human multiple myeloma immune microenvironment across disease stages.J Hematol Oncol. 2024 Nov 7;17(1):107. doi: 10.1186/s13045-024-01629-3. J Hematol Oncol. 2024. PMID: 39511632 Free PMC article.
-
Transcriptome Analysis in Mexican Adults with Acute Lymphoblastic Leukemia.Int J Mol Sci. 2024 Feb 1;25(3):1750. doi: 10.3390/ijms25031750. Int J Mol Sci. 2024. PMID: 38339034 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
