

H3K4me3 mediates uterine leiomyoma pathogenesis via neuronal processes, synapsis components, proliferation, and Wnt/β-catenin and TGF-β pathways
- PMID: 36703136
- PMCID: PMC9878797
- DOI: 10.1186/s12958-023-01060-2
H3K4me3 mediates uterine leiomyoma pathogenesis via neuronal processes, synapsis components, proliferation, and Wnt/β-catenin and TGF-β pathways
Abstract
Background: Uterine leiomyomas (UL) are the most common benign tumor in women of reproductive age. Their pathology remains unclear, which hampers the development of safe and effective treatments. Raising evidence suggests epigenetics as a main mechanism involved in tumor development. Histone modification is a key component in the epigenetic regulation of gene expression. Specifically, the histone mark H3K4me3, which promotes gene expression, is altered in many tumors. In this study, we aimed to identify if the histone modification H3K4me3 regulates the expression of genes involved in uterine leiomyoma pathogenesis.
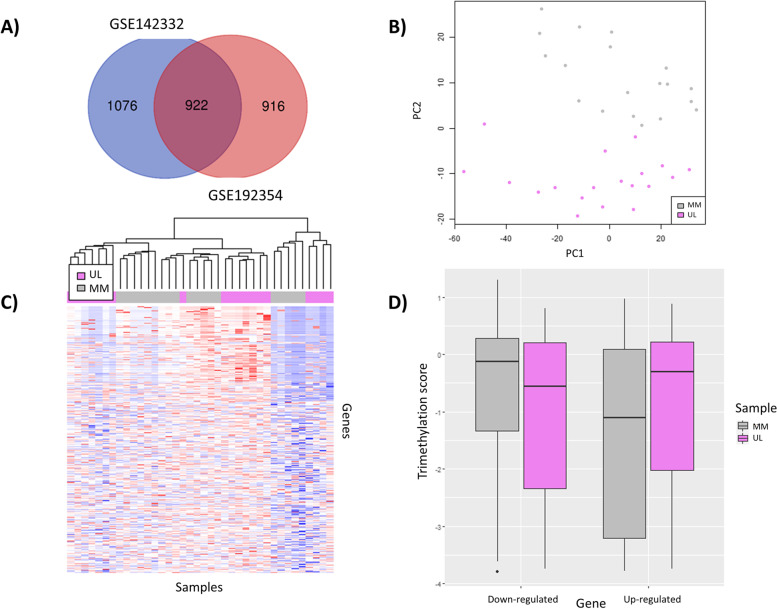
Methods: Prospective study integrating RNA-seq (n = 48) and H3K4me3 CHIP-seq (n = 19) data of uterine leiomyomas versus their adjacent myometrium. Differentially expressed genes (FDR < 0.01, log2FC > 1 or < - 1) were selected following DESeq2, edgeR, and limma analysis. Their differential methylation and functional enrichment (FDR < 0.05) were respectively analyzed with limma and ShinyGO.
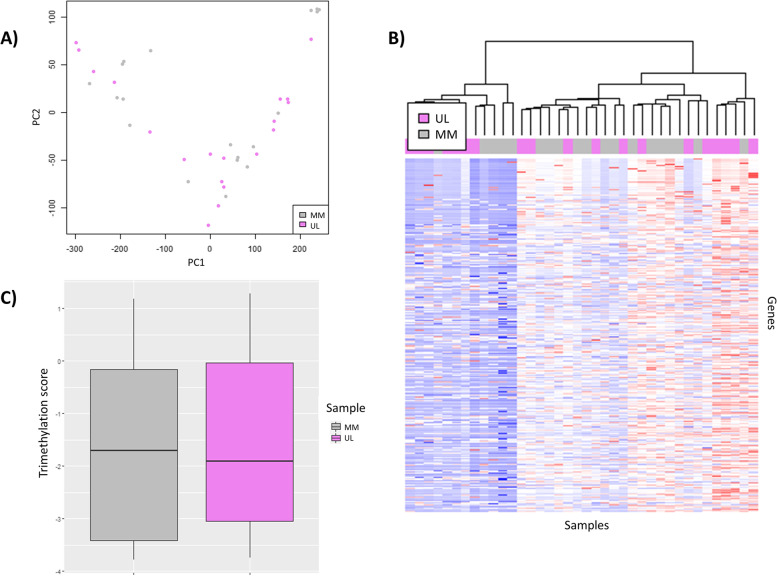
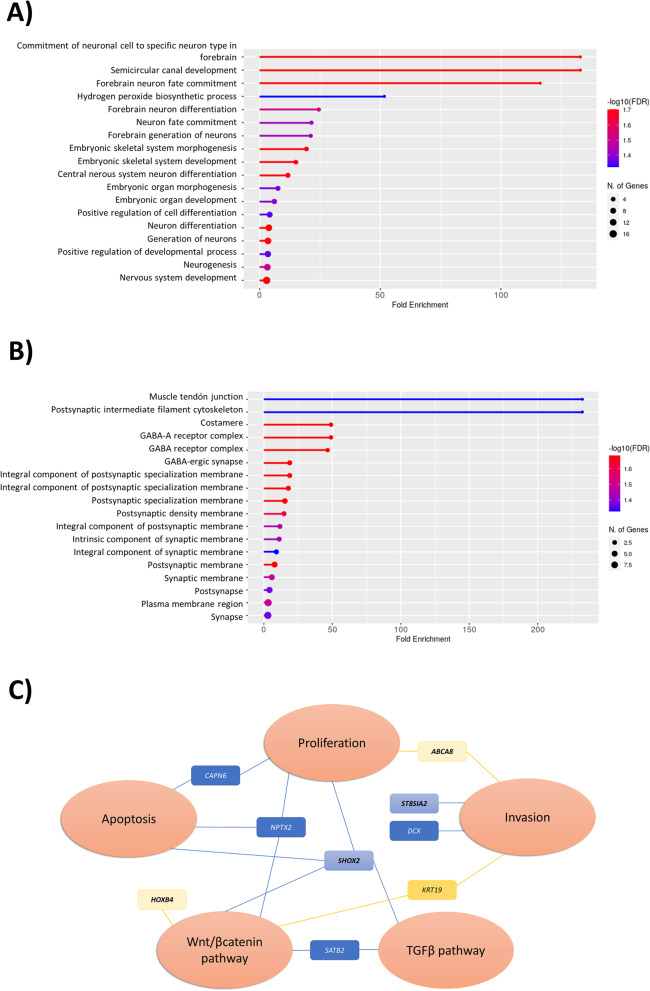
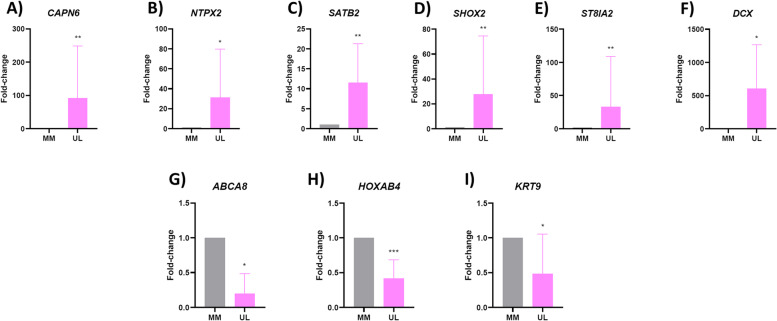
Results: CHIP-seq data showed a global suppression of H3K4me3 in uterine leiomyomas versus their adjacent myometrial tissue (p-value< 2.2e-16). Integrating CHIP-seq and RNA-seq data highlighted that transcription of 696/922 uterine leiomyoma-related differentially expressed genes (DEG) (FDR < 0.01, log2FC > 1 or < - 1) was epigenetically mediated by H3K4me3. Further, 50 genes were differentially trimethylated (FDR < 0.05), including 33 hypertrimethylated/upregulated, and 17 hypotrimethylated/downregulated genes. Functional enrichment analysis of the latter showed dysregulation of neuron-related processes and synapsis-related cellular components in uterine leiomyomas, and a literature review study of these DEG found additional implications with tumorigenesis (i.e. aberrant proliferation, invasion, and dysregulation of Wnt/β-catenin, and TGF-β pathways). Finally, SATB2, DCX, SHOX2, ST8SIA2, CAPN6, and NPTX2 proto-oncogenes were identified among the hypertrimethylated/upregulated DEG, while KRT19, ABCA8, and HOXB4 tumor suppressor genes were identified among hypotrimethylated/downregulated DEG.
Conclusions: H3K4me3 instabilities alter the expression of oncogenes and tumor suppressor genes, inducing aberrant proliferation, and dysregulated Wnt/β-catenin, and TGF-β pathways, that ultimately promote uterine leiomyoma progression. The reversal of these histone modifications may be a promising new therapeutic alternative for uterine leiomyoma patients.
Keywords: Gene expression; H3K4me3; Histone modification; Proliferation; Uterine leiomyoma; Wnt/β-catenin.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
