

What is the role of lipids in prion conversion and disease?
- PMID: 36704327
- PMCID: PMC9871914
- DOI: 10.3389/fnmol.2022.1032541
What is the role of lipids in prion conversion and disease?
Abstract
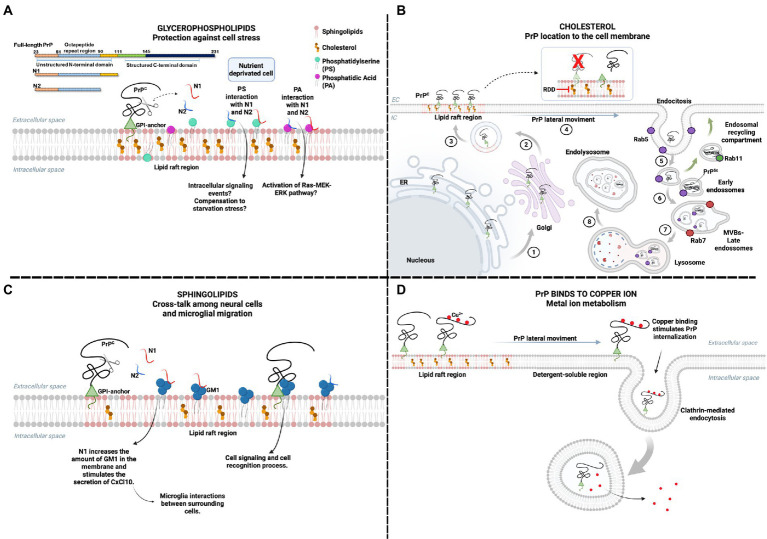
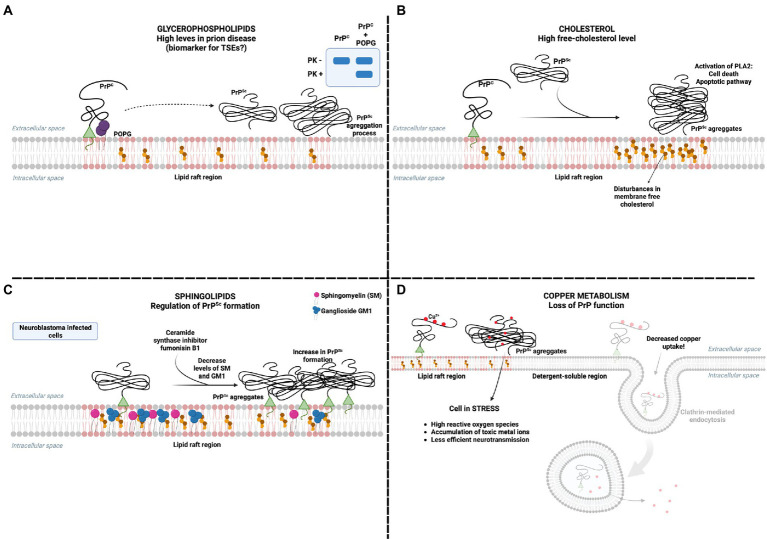
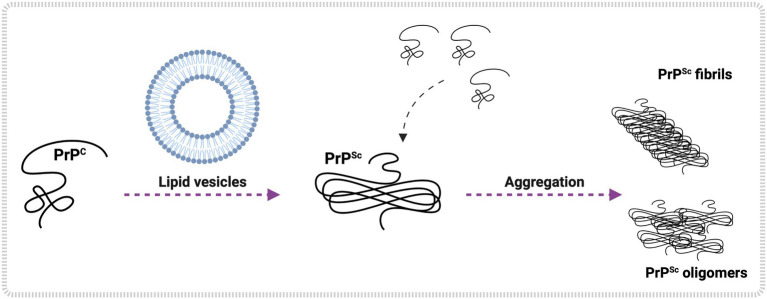
The molecular cause of transmissible spongiform encephalopathies (TSEs) involves the conversion of the cellular prion protein (PrPC) into its pathogenic form, called prion scrapie (PrPSc), which is prone to the formation of amorphous and amyloid aggregates found in TSE patients. Although the mechanisms of conversion of PrPC into PrPSc are not entirely understood, two key points are currently accepted: (i) PrPSc acts as a seed for the recruitment of native PrPC, inducing the latter's conversion to PrPSc; and (ii) other biomolecules, such as DNA, RNA, or lipids, can act as cofactors, mediating the conversion from PrPC to PrPSc. Interestingly, PrPC is anchored by a glycosylphosphatidylinositol molecule in the outer cell membrane. Therefore, interactions with lipid membranes or alterations in the membranes themselves have been widely investigated as possible factors for conversion. Alone or in combination with RNA molecules, lipids can induce the formation of PrP in vitro-produced aggregates capable of infecting animal models. Here, we discuss the role of lipids in prion conversion and infectivity, highlighting the structural and cytotoxic aspects of lipid-prion interactions. Strikingly, disorders like Alzheimer's and Parkinson's disease also seem to be caused by changes in protein structure and share pathogenic mechanisms with TSEs. Thus, we posit that comprehending the process of PrP conversion is relevant to understanding critical events involved in a variety of neurodegenerative disorders and will contribute to developing future therapeutic strategies for these devastating conditions.
Keywords: aggregation; neurodegenerative disease; prion diseases; prion protein; protein-lipid interaction.
Copyright © 2023 Alves Conceição, Assis de Lemos, Barros and Vieira.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Prion protein misfolding, strains, and neurotoxicity: an update from studies on Mammalian prions.Int J Cell Biol. 2013;2013:910314. doi: 10.1155/2013/910314. Epub 2013 Dec 24. Int J Cell Biol. 2013. PMID: 24454379 Free PMC article. Review.
-
Proteostasis unbalance in prion diseases: Mechanisms of neurodegeneration and therapeutic targets.Front Neurosci. 2022 Sep 6;16:966019. doi: 10.3389/fnins.2022.966019. eCollection 2022. Front Neurosci. 2022. PMID: 36148145 Free PMC article. Review.
-
Synthetic scrapie infectivity: interaction between recombinant PrP and scrapie brain-derived RNA.Virulence. 2015;6(2):132-44. doi: 10.4161/21505594.2014.989795. Epub 2015 Jan 13. Virulence. 2015. PMID: 25585171 Free PMC article.
-
Metal Dyshomeostasis and Their Pathological Role in Prion and Prion-Like Diseases: The Basis for a Nutritional Approach.Front Neurosci. 2017 Jan 19;11:3. doi: 10.3389/fnins.2017.00003. eCollection 2017. Front Neurosci. 2017. PMID: 28154522 Free PMC article. Review.
-
Prion protein self-interaction in prion disease therapy approaches.Vet Q. 2011 Sep;31(3):115-28. doi: 10.1080/01652176.2011.604976. Vet Q. 2011. PMID: 22029882 Review.
Cited by
-
Roles of prion proteins in mammalian development.Anim Cells Syst (Seoul). 2024 Dec 10;28(1):551-566. doi: 10.1080/19768354.2024.2436860. eCollection 2024. Anim Cells Syst (Seoul). 2024. PMID: 39664939 Free PMC article. Review.
-
Protein-lipid interactions and protein anchoring modulate the modes of association of the globular domain of the Prion protein and Doppel protein to model membrane patches.Front Bioinform. 2024 Jan 5;3:1321287. doi: 10.3389/fbinf.2023.1321287. eCollection 2023. Front Bioinform. 2024. PMID: 38250434 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials