

GRIN2B-related neurodevelopmental disorder: current understanding of pathophysiological mechanisms
- PMID: 36704660
- PMCID: PMC9873235
- DOI: 10.3389/fnsyn.2022.1090865
GRIN2B-related neurodevelopmental disorder: current understanding of pathophysiological mechanisms
Abstract
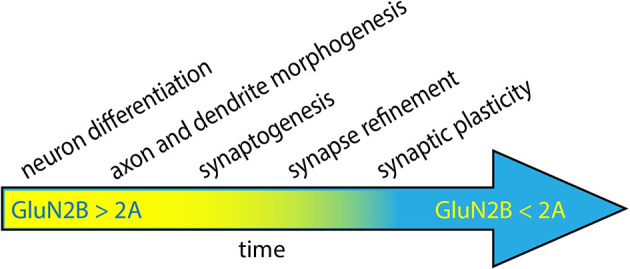
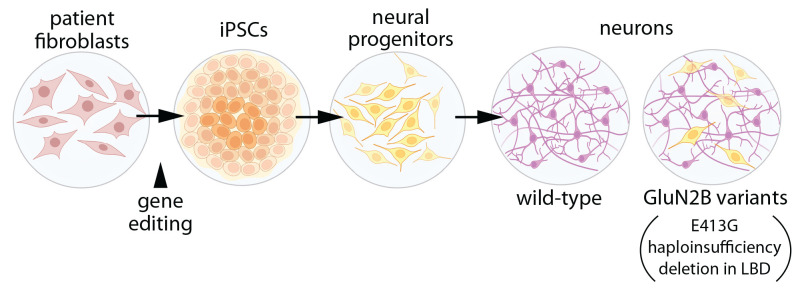
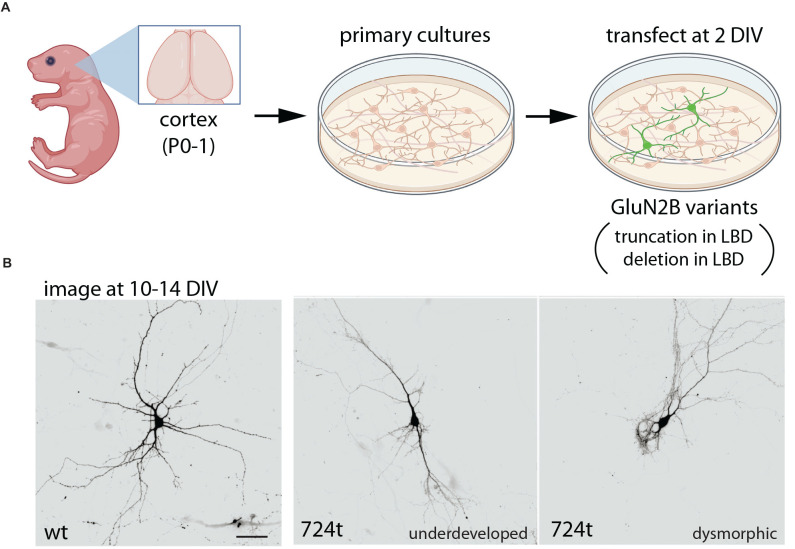
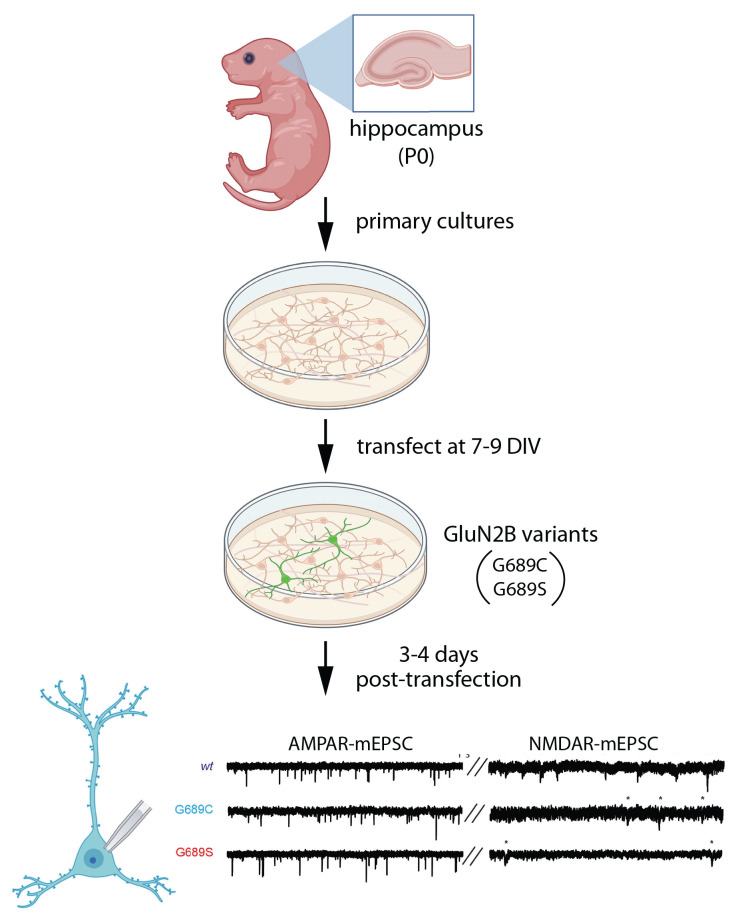
The GRIN2B-related neurodevelopmental disorder is a rare disease caused by mutations in the GRIN2B gene, which encodes the GluN2B subunit of NMDA receptors. Most individuals with GRIN2B-related neurodevelopmental disorder present with intellectual disability and developmental delay. Motor impairments, autism spectrum disorder, and epilepsy are also common. A large number of pathogenic de novo mutations have been identified in GRIN2B. However, it is not yet known how these variants lead to the clinical symptoms of the disease. Recent research has begun to address this issue. Here, we describe key experimental approaches that have been used to better understand the pathophysiology of this disease. We discuss the impact of several distinct pathogenic GRIN2B variants on NMDA receptor properties. We then critically review pivotal studies examining the synaptic and neurodevelopmental phenotypes observed when disease-associated GluN2B variants are expressed in neurons. These data provide compelling evidence that various GluN2B mutants interfere with neuronal differentiation, dendrite morphogenesis, synaptogenesis, and synaptic plasticity. Finally, we identify important open questions and considerations for future studies aimed at understanding this complex disease. Together, the existing data provide insight into the pathophysiological mechanisms that underlie GRIN2B-related neurodevelopmental disorder and emphasize the importance of comparing the effects of individual, disease-associated variants. Understanding the molecular, cellular and circuit phenotypes produced by a wide range of GRIN2B variants should lead to the identification of core neurodevelopmental phenotypes that characterize the disease and lead to its symptoms. This information could help guide the development and application of effective therapeutic strategies for treating individuals with GRIN2B-related neurodevelopmental disorder.
Keywords: GRIN2B; GluN2B (NMDA receptor subunit NR2B); NMDAR (NMDA receptor); autism (ASD); dendrite development; disease variants; neuron development; synapse development.
Copyright © 2023 Sabo, Lahr, Offer, Weekes and Sceniak.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
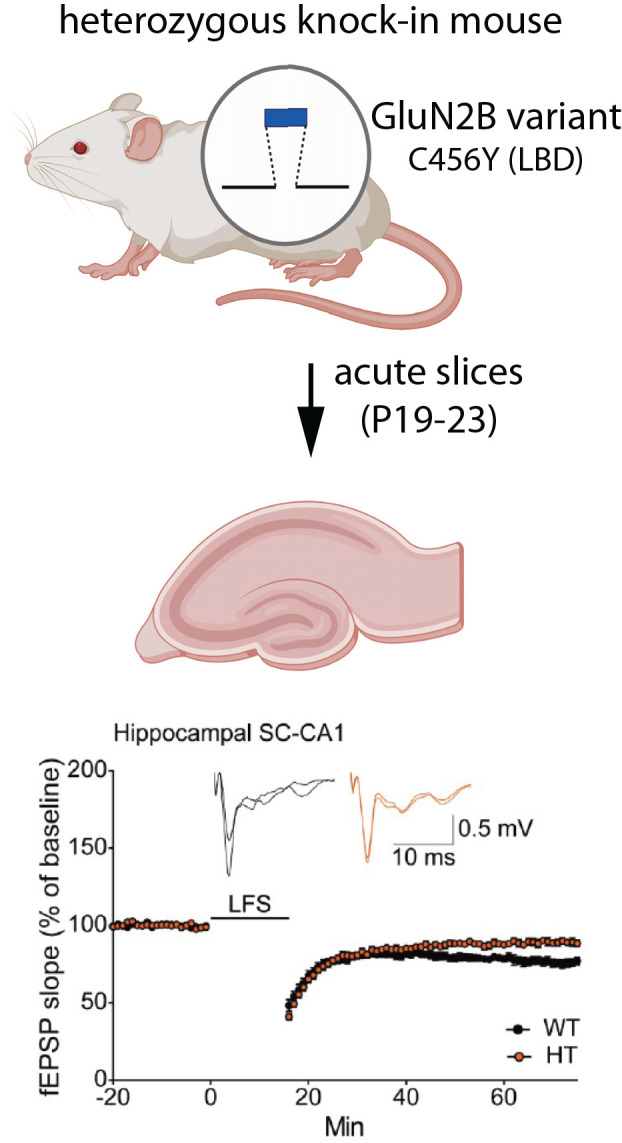
Similar articles
-
Human GRIN2B variants in neurodevelopmental disorders.J Pharmacol Sci. 2016 Oct;132(2):115-121. doi: 10.1016/j.jphs.2016.10.002. Epub 2016 Oct 19. J Pharmacol Sci. 2016. PMID: 27818011 Free PMC article. Review.
-
Characterization of Mice Carrying a Neurodevelopmental Disease-Associated GluN2B(L825V) Variant.J Neurosci. 2024 Jul 31;44(31):e2291232024. doi: 10.1523/JNEUROSCI.2291-23.2024. J Neurosci. 2024. PMID: 38926089 Free PMC article.
-
Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification.Brain Sci. 2022 Jun 15;12(6):789. doi: 10.3390/brainsci12060789. Brain Sci. 2022. PMID: 35741674 Free PMC article.
-
Disruption of grin2B, an ASD-associated gene, produces social deficits in zebrafish.Mol Autism. 2022 Sep 22;13(1):38. doi: 10.1186/s13229-022-00516-3. Mol Autism. 2022. PMID: 36138431 Free PMC article.
-
Distinct roles of GRIN2A and GRIN2B variants in neurological conditions.F1000Res. 2019 Nov 20;8:F1000 Faculty Rev-1940. doi: 10.12688/f1000research.18949.1. eCollection 2019. F1000Res. 2019. PMID: 31807283 Free PMC article. Review.
Cited by
-
Limb-Clasping Response in NMDA Receptor Palmitoylation-Deficient Mice.Mol Neurobiol. 2024 Nov;61(11):9125-9135. doi: 10.1007/s12035-024-04166-9. Epub 2024 Apr 9. Mol Neurobiol. 2024. PMID: 38592586 Free PMC article.
-
Neuroimaging to Genotype: Delineating the Spectrum of Disorders With Deficient Myelination in the Indian Population.Am J Med Genet A. 2025 Mar;197(3):e63914. doi: 10.1002/ajmg.a.63914. Epub 2024 Oct 29. Am J Med Genet A. 2025. PMID: 39470296 Free PMC article.
-
The Evolving Landscape of Functional Models of Autism Spectrum Disorder.Cells. 2025 Jun 16;14(12):908. doi: 10.3390/cells14120908. Cells. 2025. PMID: 40558535 Free PMC article. Review.
-
AMPA and NMDA Receptors in Hippocampus of Rats with Fluoride-Induced Cognitive Decline.Int J Mol Sci. 2024 Nov 2;25(21):11796. doi: 10.3390/ijms252111796. Int J Mol Sci. 2024. PMID: 39519348 Free PMC article.
-
Genomic evidence for the suitability of Göttingen Minipigs with a rare seizure phenotype as a model for human epilepsy.Neurogenetics. 2024 Apr;25(2):103-117. doi: 10.1007/s10048-024-00750-2. Epub 2024 Feb 21. Neurogenetics. 2024. PMID: 38383918 Free PMC article.
References
-
- Alavian-Ghavanini A., Lin P.-I., Lind P. M., Rimfors S. R., Halin Lejonklou M., Dunder L., et al. . (2018). Prenatal bisphenol A exposure is linked to epigenetic changes in glutamate receptor subunit gene grin2b in female rats and humans. Sci. Rep. 8:11315. 10.1038/s41598-018-29732-9 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous