

Infancy-onset diabetes caused by de-regulated AMPylation of the human endoplasmic reticulum chaperone BiP
- PMID: 36704923
- PMCID: PMC9994480
- DOI: 10.15252/emmm.202216491
Infancy-onset diabetes caused by de-regulated AMPylation of the human endoplasmic reticulum chaperone BiP
Abstract
Dysfunction of the endoplasmic reticulum (ER) in insulin-producing beta cells results in cell loss and diabetes mellitus. Here we report on five individuals from three different consanguineous families with infancy-onset diabetes mellitus and severe neurodevelopmental delay caused by a homozygous p.(Arg371Ser) mutation in FICD. The FICD gene encodes a bifunctional Fic domain-containing enzyme that regulates the ER Hsp70 chaperone, BiP, via catalysis of two antagonistic reactions: inhibitory AMPylation and stimulatory deAMPylation of BiP. Arg371 is a conserved residue in the Fic domain active site. The FICDR371S mutation partially compromises BiP AMPylation in vitro but eliminates all detectable deAMPylation activity. Overexpression of FICDR371S or knock-in of the mutation at the FICD locus of stressed CHO cells results in inappropriately elevated levels of AMPylated BiP and compromised secretion. These findings, guided by human genetics, highlight the destructive consequences of de-regulated BiP AMPylation and raise the prospect of tuning FICD's antagonistic activities towards therapeutic ends.
Keywords: diabetes mellitus; endoplasmic reticulum chaperone; mutation; nucleotidyltransferases; post-translational.
© 2023 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
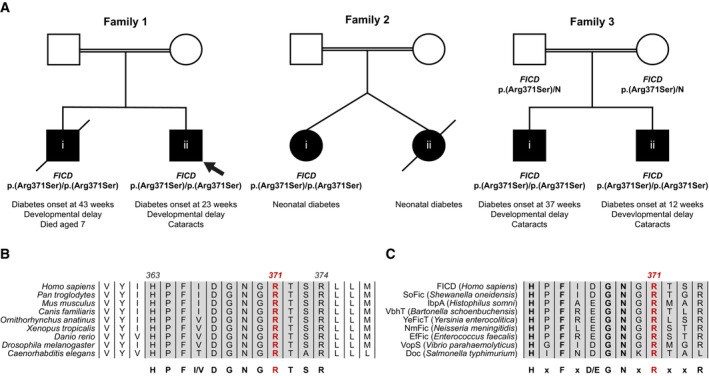
- A
Partial pedigrees of three families found to bear this rare genetic variant. Where available, information pertaining to the FICD genotype and the phenotype of affected individuals are shown. The first individual identified to carry this homozygous mutation (the proband, family member 1ii) is indicated with an arrow.
- B, C
Arg371 is located within the catalytic Fic motif and is highly conserved both across metazoan FICD orthologues (B) and across the wider Fic and Doc protein superfamily (C). The Fic motif is highlighted in grey, and residue numbering corresponds to human FICD.
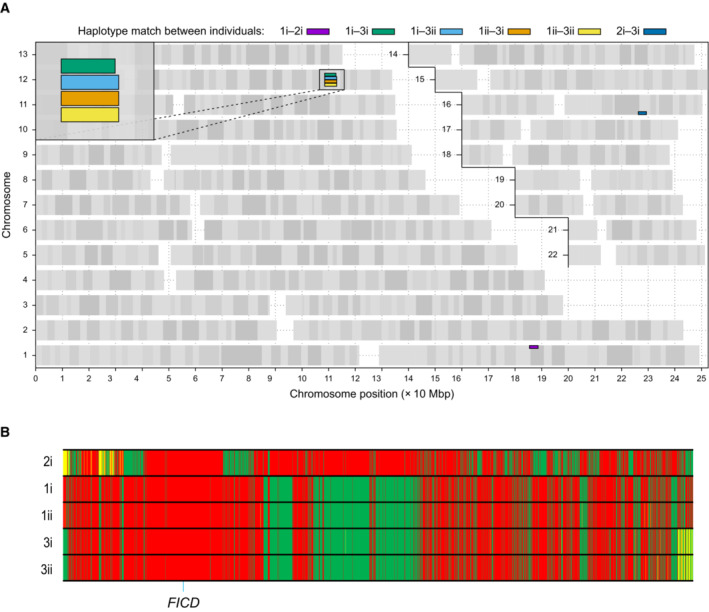
- A
Graphical representation of genome‐wide shared genomic segments larger than 3 Mbp in the five individuals with FICD homozygous mutations. Each colour bar represents a haplotype shared by two individuals (across all four copies of the same chromosome), labelled according to Fig 1A. The four rectangles on the long arm of chromosome 12 (inset) show that the four individuals in Families 1 and 3 share a haplotype [approximate coordinates chr12(hg19):108512514–113023258] including the FICD gene [chr12(hg19):108909051–108913380], but none of them shares a haplotype with the patient in Family 2 (individual 2i) —hence the lack of annotated purple or dark blue bars. Note, genotype information was not available for family member 2ii.
- B
Genotype calls for 2,735 single nucleotide, not multiallelic variants, located on chr12:(hg19):108500000–113100000 in the five individuals with the FICD homozygous p.(Arg371Ser) mutation. Only variants where at least one of the five patients carries the alternative allele are shown. Variants with a coverage of < 15 reads and an allele balance for heterozygous calls < 0.25 were removed. The position of the three variants in the FICD locus (including the pathogenic p.(Arg371Ser) variant) is indicated at the bottom of the graph. Green = homozygous for the reference allele, Yellow = heterozygous, Red = homozygous for the alternative allele. Note the similarity in variant pattern in affected individuals from families 1 & 3 and the dissimilarity with the affected member of family 2.
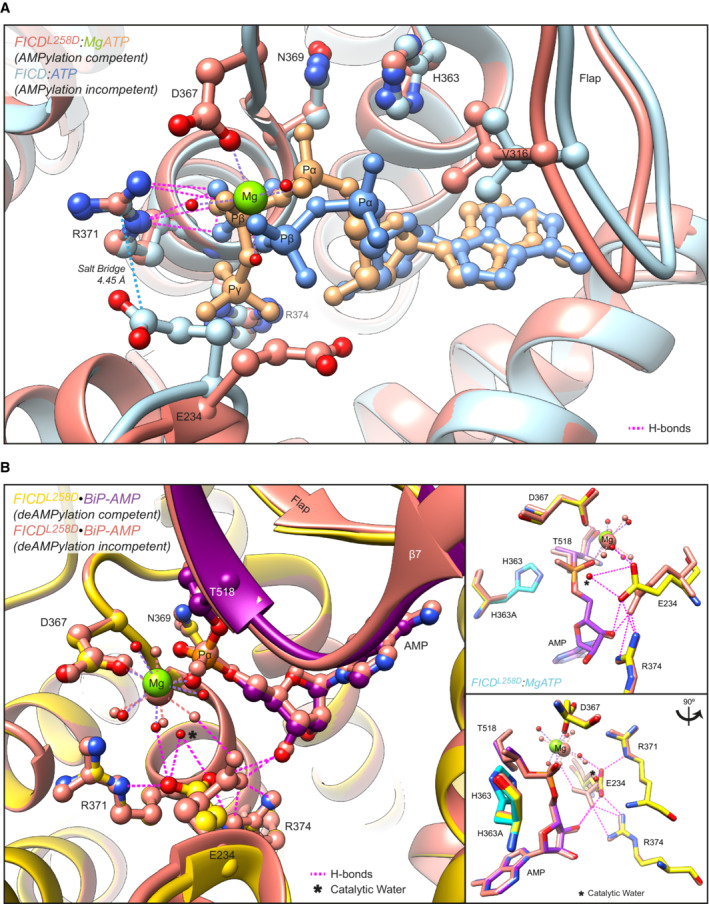
- A
Structures of monomeric (FICDL258D) and dimeric FICD proteins, crystallised in the presence of a large excess of magnesium cations and ATP, are superposed (PDB 6I7K and 6I7G, respectively). The former engages MgATP in an AMPylation competent conformation and the latter engages (only) ATP in an AMPylation incompetent conformation (Perera et al, 2019). Arg371 forms hydrogen bonds with both nucleotides (pink dashed lines). Arg371's interaction with the β‐phosphate of MgATP is also likely to help stabilise the AMPylation transition state. A potential long range ionic interaction between Arg371 and Glu234 in the non‐competent conformation is also annotated.
- B
Two alternative states of FICD's active site engaged with its deAMPylation substrate, BiP‐AMP. Glu234 can exist in at least two conformations. In the deAMPylation competent conformation, Glu234 correctly orientates a catalytic water molecule (*) for in‐line nucleophilic attack into the backside of the BiP‐AMP phosphodiester bond (PDB 7B7Z). In the deAMPylation incompetent conformation Glu234 points away from the position of this putative catalytic water (PDB 7B80) (Perera et al, 2021). Hydrogen bonds formed by Glu234 are annotated in both cases (pink dashed lines). Arg371 interacts with Glu234 only when it resides in a deAMPylation competent conformation. Insets, reduced view of the superposed active sites, shown in orthogonal views, additionally overlaid with the catalytic His363 residues from PDB 6I7K (cyan).
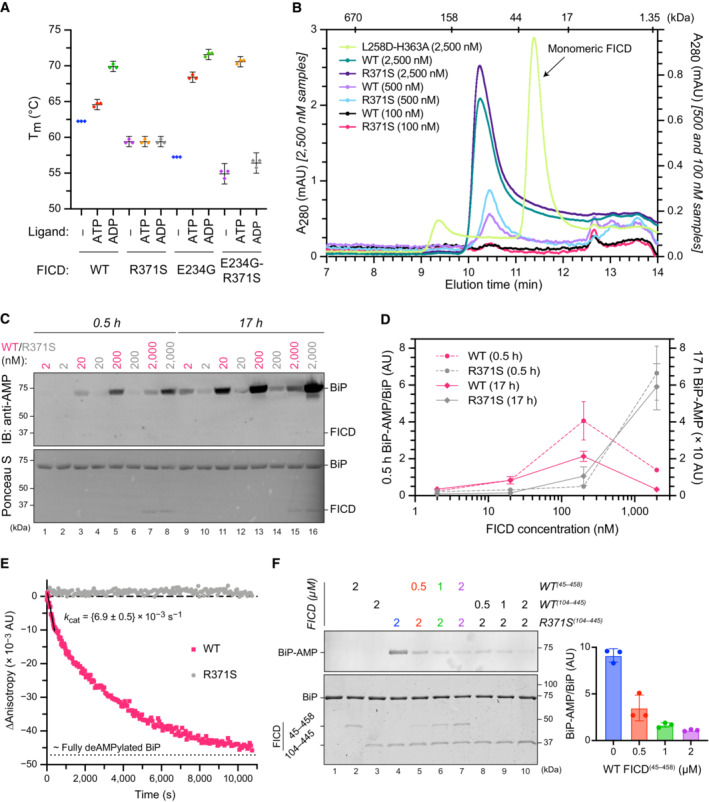
- A
Plot of the principal melting temperatures (T ms) of wild‐type FICD (WT) and FICDR371S in the presence or absence of nucleotide ligands and/or FICD's gatekeeper Glu234 residue, derived from differential scanning fluorimetry (DSF). The mean T m ± 95% confidence interval (CI) is displayed, from n = 3 independent experiments. Arg371Ser mutation slightly destabilises the apo enzyme and significantly reduces the ability of ATP and ADP to stabilise FICD. Glu234Gly mutation partially restores ATP (but not ADP) stabilisation of FICDE234G‐R371S (see Fig EV2A).
- B
Size exclusion chromatography confirms that the oligomerisation tendency of FICDR371S is unperturbed relative to wild‐type FICD. Across a range of assessable enzyme concentrations both FICDs elute as predominantly dimeric species. The elution profile of a monomeric FICDL258D‐H363A mutant is also shown for reference, alongside the elution times of molecular weight standards.
- C
Representative immunoblot comparing the ability of wild‐type and Arg371Ser FICD (at a wide range of enzyme concentrations) to catalyse the accumulation of BiP‐AMP in the presence of physiological ATP concentrations (5 mM). At low enzyme concentrations (and early time points) the AMPylation defect imposed by Arg371Ser is manifest. At higher enzyme concentrations where the deAMPylation activity of the wild‐type enzyme begins to dominate, the residual AMPylation activity of FICDR371S results in a greater accumulation of BiP‐AMP (relative to the same concentration of wild‐type enzyme).
- D
Quantification of the AMPylated BiP signals, relative to total BiP, from experiments as in (C) (mean values ± SEM, n = 4, biological replicates). Figure EV2B displays the values from all four repetitions.
- E
A fluorescence polarisation‐based deAMPylation assay highlighting the lack of activity catalysed by FICDR371S. Enzyme‐saturating concentrations of BiP‐AMP were provided as deAMPylation substrate. No enzymatic activity of 10 μM (grey trace, shown) or 20 μM enzyme FICDR371S was detectable. The estimated fluorescence anisotropy value of a fully deAMPylated BiP sample is derived from a heuristic fitting of a single exponential decay curve. The k cat value for wild‐type FICD was calculated from n = 3 biological replicates (mean ± SEM).
- F
Representative immunoblot comparing the accumulation of BiP‐AMP in reactions constituted with the indicated concentrations of wild‐type and Arg371Ser FICD, performed as in (C) (upper left panel). The lower left panel is a Coomassie‐stained gel of the reaction constituents. An N‐terminally extended version of the wild‐type enzyme (residues 45–458 was used in lanes 2 and 5–7, to distinguish its migration on the gel from the mutant (residues 104–445). The bar diagram provides quantification of the AMPylated BiP signals relative to total BiP (from n = 3, biological replicates, mean values ± SEM).
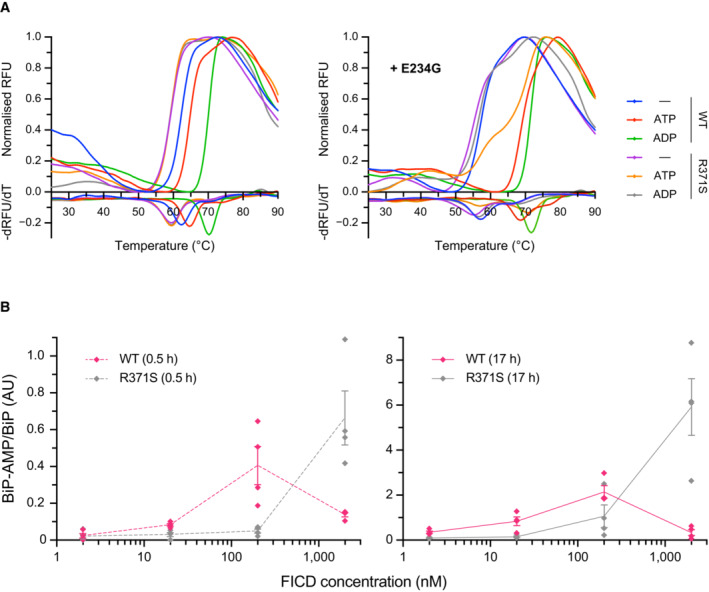
- A
Representative (of experiments reproduced three time), normalised differential scanning fluorimetry (DSF) melt curves of the indicated FICD proteins (1 μM) in presence and absence of nucleotide (2.5 mM), shown above their corresponding negative first‐derivatives. Note, FICDE234G‐R371S displays a non‐uniform T m shift in response to ATP.
- B
Quantification of the AMPylated BiP signals relative to total BiP of experiments displaying the same data in Fig 3C (mean values ± SEM, n = 4, biological replicates), but split into the two time points of the experiment and presenting the values from all four replicates.
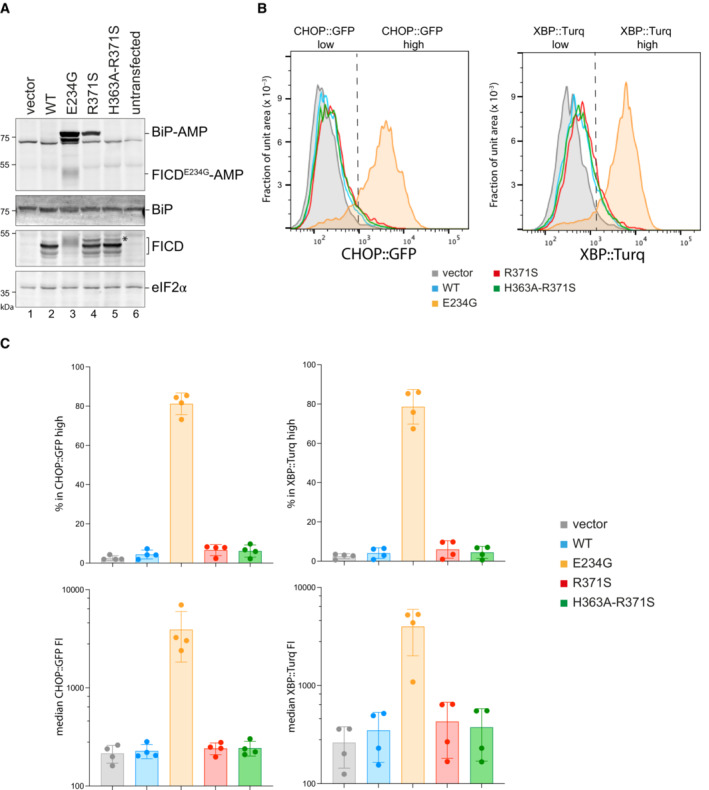
- A
Immunoblots of AMPylated proteins detected with a pan‐AMP antibody, BiP, FICD and eIF2α (a loading control) in lysates of FICD ∆ CHO cells transiently transfected with plasmids encoding the indicated derivatives of FICD. Signals corresponding to AMPylated BiP, auto‐AMPylated FICDE234G, total BiP, FICD and eIF2α are indicated. The novel species reactive with the anti‐FICD antiserum in lysates of cells transfected with plasmids expressing FICDR371S (*) likely reflects a glycosylated isoform arising from the creation of a new glycosylation site at Asn369.
- B
Histograms of Integrated Stress Response reporter CHOP::GFP and XBP::Turquoise (XBP::Turq, a reporter of the IRE1 branch of the UPR) fluorescence in FICD ∆ CHO dual reporter cells transfected with expression vectors as in (A). Transfected cells were gated for mCherry positivity, a marker carried on the expression plasmid in trans to the indicated FICD effector.
- C
Quantification of the data from repeats of the experiment as shown in (B). The upper two graphs show the percent of cells with high CHOP::GFP or high XBP::Turq for each transfection. The lower panel provides quantitation of the median fluorescence intensity for each reporter. Mean ± SD, n = 4, biological replicates.
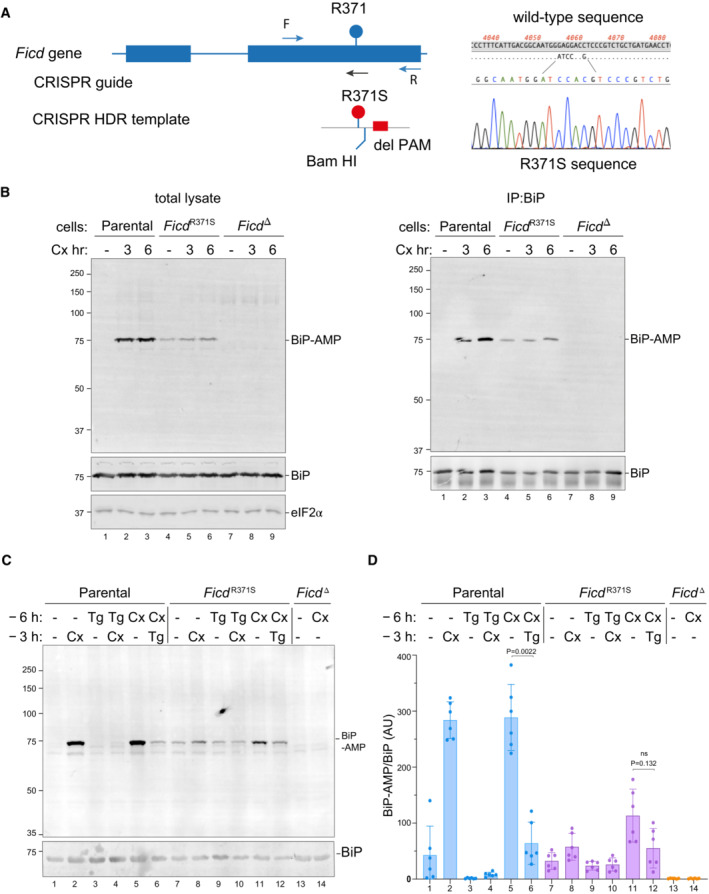
- A
On the left is a schema of the Chinese Hamster Ovary cell FICD locus. Exons depicted as blue boxes, introns as lines. Forward (F) and reverse (R) primers used to genotype the locus and the guide RNA that directs the Cas9 nuclease (CRISPR guide) are depicted by arrows. A schema of the repair template is provided below, with the Arg371Ser mutation, the silent BamHI site and silent PAM‐destroying features noted. To the right is the sequence of the locus before and after recombination and a sequencing trace of the locus in the mutant FICD R371S derivative cell line.
- B
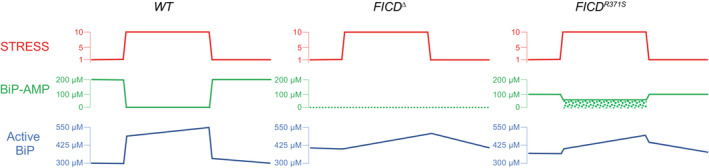
On the left are immunoblots of AMPylated proteins detected with pan‐AMP, BiP and eIF2α (a loading control) reactive antibodies in lysates of cells of the indicated genotype. The cells were untreated or exposed to the protein synthesis inhibitor, cycloheximide (Cx, 100 μg/ml) for the indicated time. In the panel on the right are immunoblots of samples immunoprecipitated with a BiP‐specific antiserum from the lysates of the samples shown on the left.
- C
Immunoblots as in (B) in lysates of cells of the indicated genotype. The cells were untreated or exposed to the protein synthesis inhibitor, cycloheximide (Cx, 100 μg/ml) and/or thapsigargin (Tg) for the indicated periods prior to harvest.
- D
Quantification of ratio of the BiP‐AMP to total BiP signal (expressed in arbitrary units) of repeats of the experiment shown in (C). Mean ± SD of the BiP‐AMP signal normalised to total BiP signal from the same samples are depicted (P values by two tailed t‐test, n = 6, biological replicates).
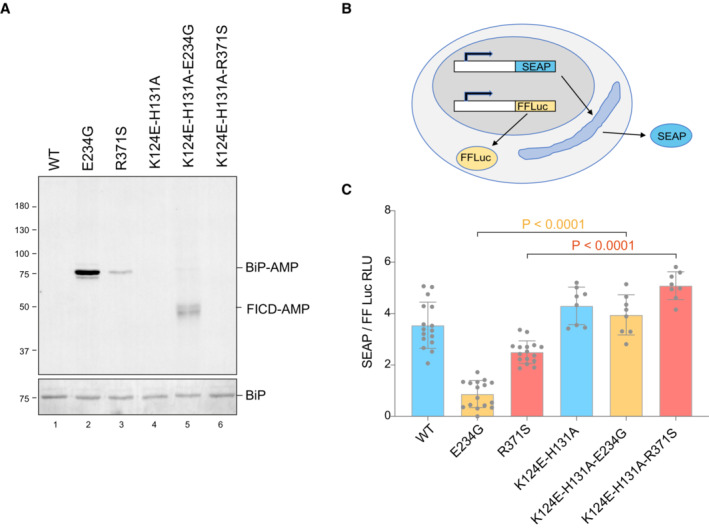
- A
Representative immunoblot of AMPylated proteins in FICD ∆ CHO cells transiently transfected with expression plasmids encoding the indicated FICD enzymes (replicated three times). Note that the hyperactive E234G mutant bearing the K124E‐H131A mutations that compromise its ability to interact with BiP, is nonetheless consistently auto‐AMPylated.
- B
Schema of the assay used to measure the effect of FICD on secretion.
- C
Bar diagram of the amount of alkaline phosphatase (normalised to cytoplasmic luciferase [a transfection marker]) secreted from FICD ∆ CHO cells co‐expressing the indicated FICD enzymes. The K124E‐H131A mutations that interfere with engagement of BiP as a substrate reverse the secretion defect imposed by the hyperactive FICD enzymes (Mean ± SD, P values by ANOVA with Šídák's multiple comparisons test, n = 8–16 [biological replicates]).
Similar articles
-
An oligomeric state-dependent switch in the ER enzyme FICD regulates AMPylation and deAMPylation of BiP.EMBO J. 2019 Oct 4;38(21):e102177. doi: 10.15252/embj.2019102177. Epub 2019 Sep 18. EMBO J. 2019. PMID: 31531998 Free PMC article.
-
Structures of a deAMPylation complex rationalise the switch between antagonistic catalytic activities of FICD.Nat Commun. 2021 Aug 18;12(1):5004. doi: 10.1038/s41467-021-25076-7. Nat Commun. 2021. PMID: 34408154 Free PMC article.
-
FICD acts bifunctionally to AMPylate and de-AMPylate the endoplasmic reticulum chaperone BiP.Nat Struct Mol Biol. 2017 Jan;24(1):23-29. doi: 10.1038/nsmb.3337. Epub 2016 Dec 5. Nat Struct Mol Biol. 2017. PMID: 27918543 Free PMC article.
-
AMPylation and Endoplasmic Reticulum Protein Folding Homeostasis.Cold Spring Harb Perspect Biol. 2023 Mar 1;15(3):a041265. doi: 10.1101/cshperspect.a041265. Cold Spring Harb Perspect Biol. 2023. PMID: 36041787 Free PMC article. Review.
-
FIC proteins: from bacteria to humans and back again.Pathog Dis. 2018 Mar 1;76(2). doi: 10.1093/femspd/fty012. Pathog Dis. 2018. PMID: 29617857 Review.
Cited by
-
Diabetes mellitus and the key role of endoplasmic reticulum stress in pancreatic β cells.Nat Rev Endocrinol. 2025 Sep;21(9):546-563. doi: 10.1038/s41574-025-01129-5. Epub 2025 Jun 4. Nat Rev Endocrinol. 2025. PMID: 40467970 Review.
-
Pre-clinical model of dysregulated FicD AMPylation causes diabetes by disrupting pancreatic endocrine homeostasis.Mol Metab. 2025 May;95:102120. doi: 10.1016/j.molmet.2025.102120. Epub 2025 Mar 10. Mol Metab. 2025. PMID: 40073934 Free PMC article.
-
Small-Molecule FICD Inhibitors Suppress Endogenous and Pathologic FICD-Mediated Protein AMPylation.ACS Chem Biol. 2025 Apr 18;20(4):880-895. doi: 10.1021/acschembio.4c00847. Epub 2025 Mar 4. ACS Chem Biol. 2025. PMID: 40036289 Free PMC article.
-
Small molecule FICD inhibitors suppress endogenous and pathologic FICD-mediated protein AMPylation.bioRxiv [Preprint]. 2024 Jul 16:2024.07.13.603377. doi: 10.1101/2024.07.13.603377. bioRxiv. 2024. Update in: ACS Chem Biol. 2025 Apr 18;20(4):880-895. doi: 10.1021/acschembio.4c00847. PMID: 39071275 Free PMC article. Updated. Preprint.
-
Visual impairment cell non-autonomously dysregulates brain-wide proteostasis.bioRxiv [Preprint]. 2023 Oct 23:2023.10.19.563166. doi: 10.1101/2023.10.19.563166. bioRxiv. 2023. PMID: 37961457 Free PMC article. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
