

Nanoscale structural organization and stoichiometry of the budding yeast kinetochore
- PMID: 36705601
- PMCID: PMC9929930
- DOI: 10.1083/jcb.202209094
Nanoscale structural organization and stoichiometry of the budding yeast kinetochore
Abstract
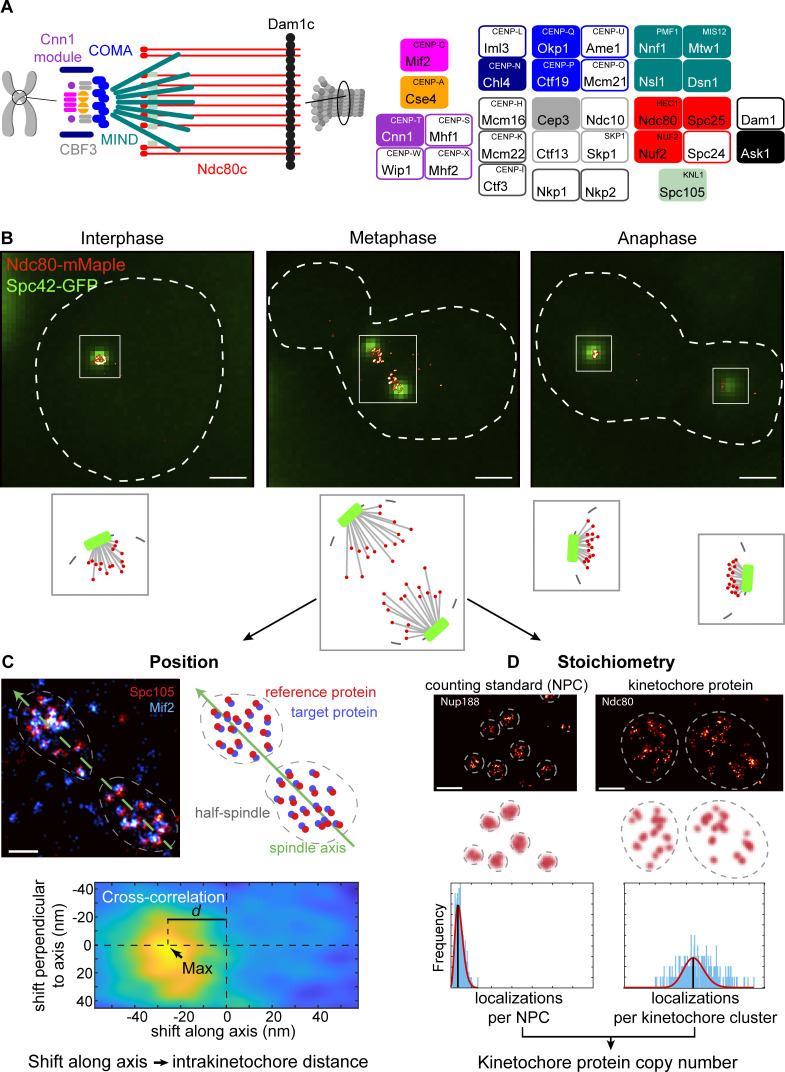
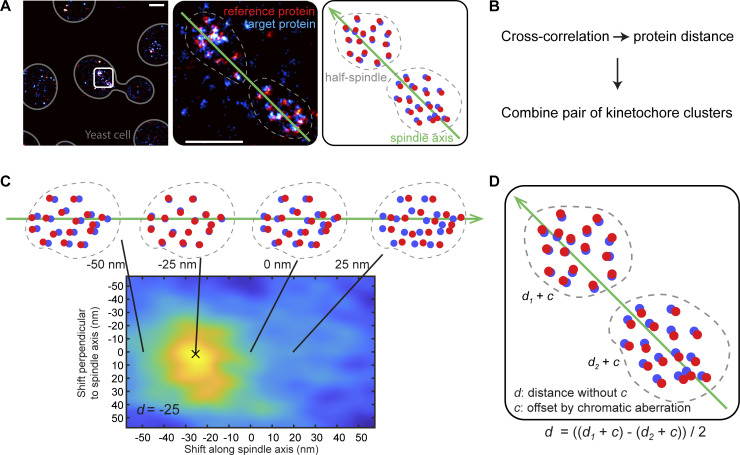
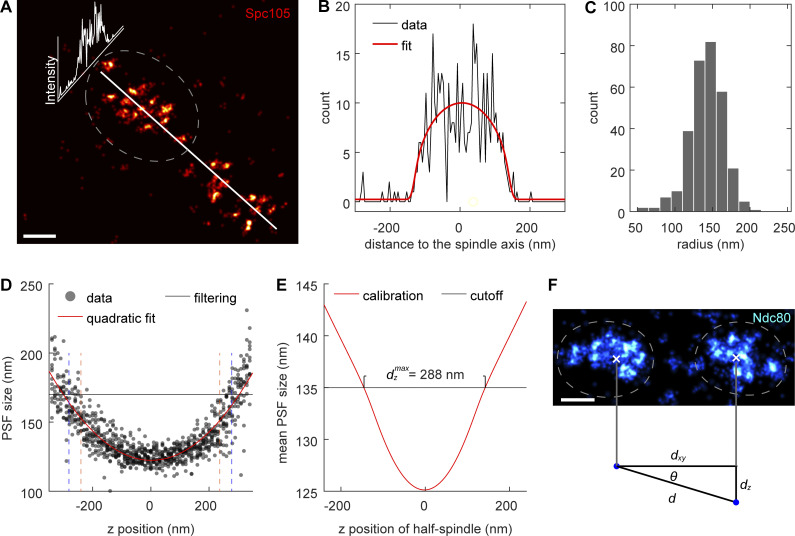
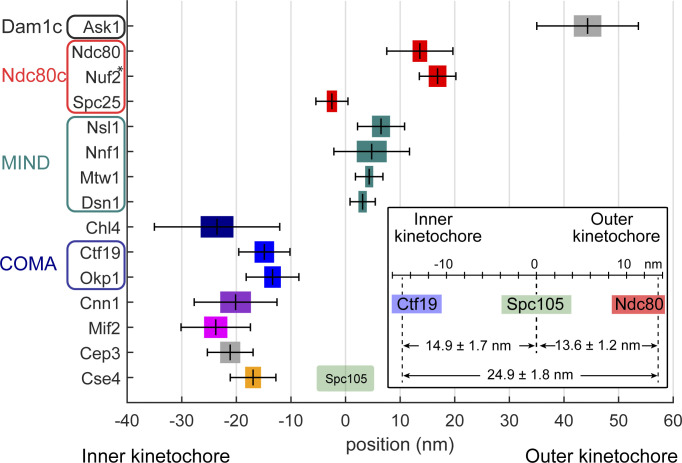
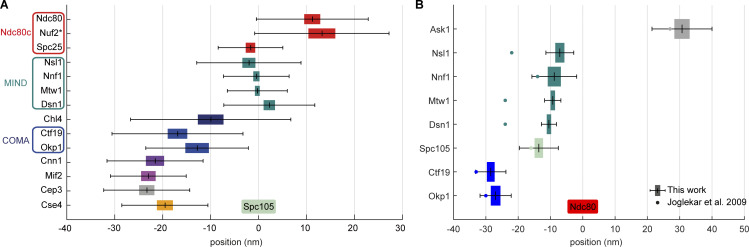
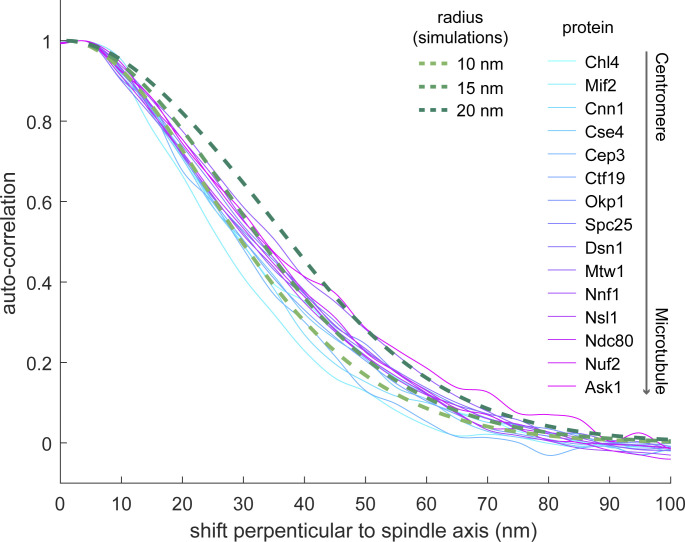
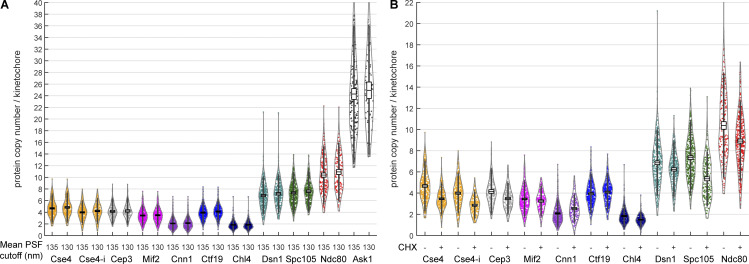
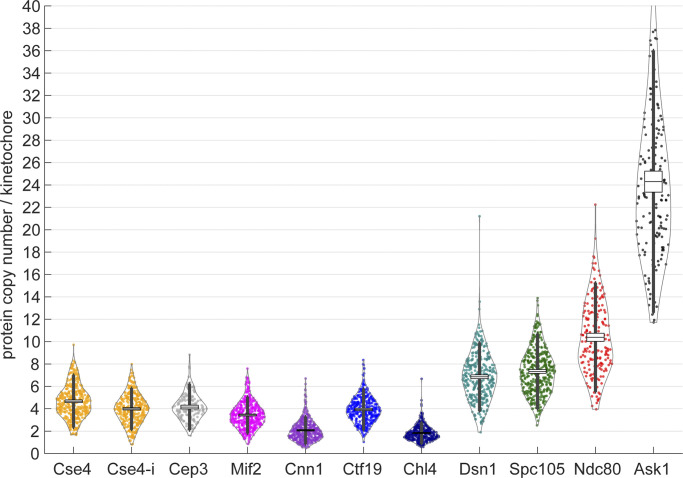
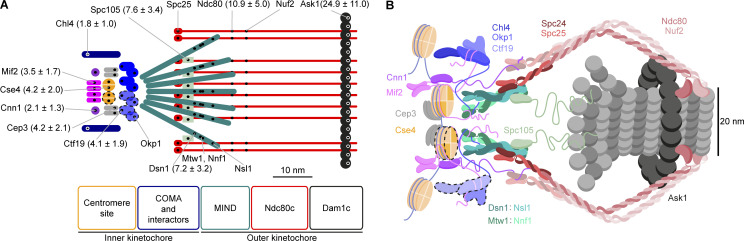
Proper chromosome segregation is crucial for cell division. In eukaryotes, this is achieved by the kinetochore, an evolutionarily conserved multiprotein complex that physically links the DNA to spindle microtubules and takes an active role in monitoring and correcting erroneous spindle-chromosome attachments. Our mechanistic understanding of these functions and how they ensure an error-free outcome of mitosis is still limited, partly because we lack a complete understanding of the kinetochore structure in the cell. In this study, we use single-molecule localization microscopy to visualize individual kinetochore complexes in situ in budding yeast. For major kinetochore proteins, we measured their abundance and position within the metaphase kinetochore. Based on this comprehensive dataset, we propose a quantitative model of the budding yeast kinetochore. While confirming many aspects of previous reports based on bulk imaging, our results present a unifying nanoscale model of the kinetochore in budding yeast.
© 2023 Cieslinski et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures
Similar articles
-
The composition, functions, and regulation of the budding yeast kinetochore.Genetics. 2013 Aug;194(4):817-46. doi: 10.1534/genetics.112.145276. Genetics. 2013. PMID: 23908374 Free PMC article. Review.
-
Slk19 clusters kinetochores and facilitates chromosome bipolar attachment.Mol Biol Cell. 2013 Mar;24(5):566-77. doi: 10.1091/mbc.E12-07-0552. Epub 2013 Jan 2. Mol Biol Cell. 2013. PMID: 23283988 Free PMC article.
-
A Gradient in Metaphase Tension Leads to a Scaled Cellular Response in Mitosis.Dev Cell. 2019 Apr 8;49(1):63-76.e10. doi: 10.1016/j.devcel.2019.01.018. Epub 2019 Feb 21. Dev Cell. 2019. PMID: 30799228 Free PMC article.
-
Kinetochore-microtubule error correction for biorientation: lessons from yeast.Biochem Soc Trans. 2024 Feb 28;52(1):29-39. doi: 10.1042/BST20221261. Biochem Soc Trans. 2024. PMID: 38305688 Free PMC article. Review.
-
Stochastic Modeling Yields a Mechanistic Framework for Spindle Attachment Error Correction in Budding Yeast Mitosis.Cell Syst. 2017 Jun 28;4(6):645-650.e5. doi: 10.1016/j.cels.2017.05.003. Epub 2017 Jun 7. Cell Syst. 2017. PMID: 28601560 Free PMC article.
Cited by
-
Unraveling the kinetochore nanostructure in Schizosaccharomyces pombe using multi-color SMLM imaging.J Cell Biol. 2023 Apr 3;222(4):e202209096. doi: 10.1083/jcb.202209096. Epub 2023 Jan 27. J Cell Biol. 2023. PMID: 36705602 Free PMC article.
-
Build and operation of a custom 3D, multicolor, single-molecule localization microscope.Nat Protoc. 2024 Aug;19(8):2467-2525. doi: 10.1038/s41596-024-00989-x. Epub 2024 May 3. Nat Protoc. 2024. PMID: 38702387 Review.
-
Super-Resolution Microscopy of the Synaptonemal Complex within the Caenorhabditis elegans Germline.J Vis Exp. 2022 Sep 13;(187):10.3791/64363. doi: 10.3791/64363. J Vis Exp. 2022. PMID: 36190293 Free PMC article.
-
Structural mechanism of outer kinetochore Dam1-Ndc80 complex assembly on microtubules.Science. 2023 Dec 8;382(6675):1184-1190. doi: 10.1126/science.adj8736. Epub 2023 Dec 7. Science. 2023. PMID: 38060647 Free PMC article.
-
The centromere bottlebrush requires a multi-microtubule attachment.Mol Biol Cell. 2025 Jun 1;36(6):ar70. doi: 10.1091/mbc.E25-02-0050. Epub 2025 Apr 23. Mol Biol Cell. 2025. PMID: 40266738
References
-
- Abbe, E. 1873. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Für Mikrosk. Anat. 9:413–418
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
