

Factor XII contributes to thrombotic complications and vaso-occlusion in sickle cell disease
- PMID: 36706361
- PMCID: PMC10122107
- DOI: 10.1182/blood.2022017074
Factor XII contributes to thrombotic complications and vaso-occlusion in sickle cell disease
Abstract
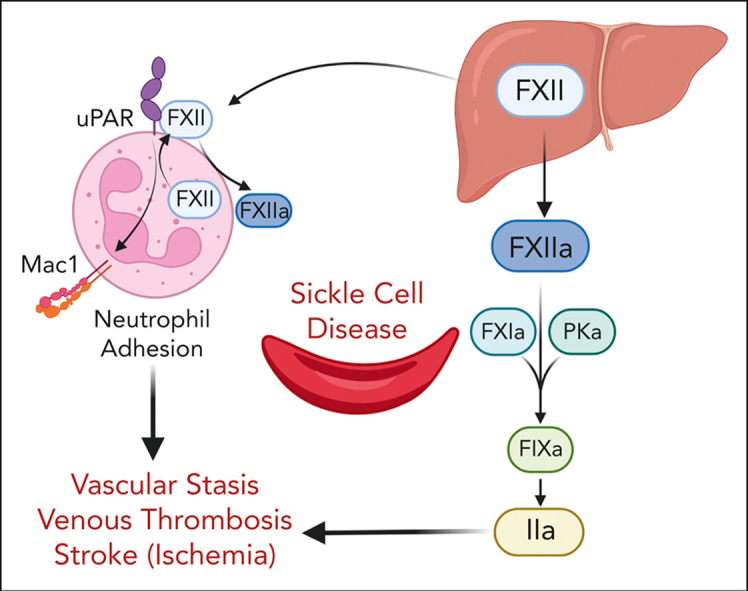
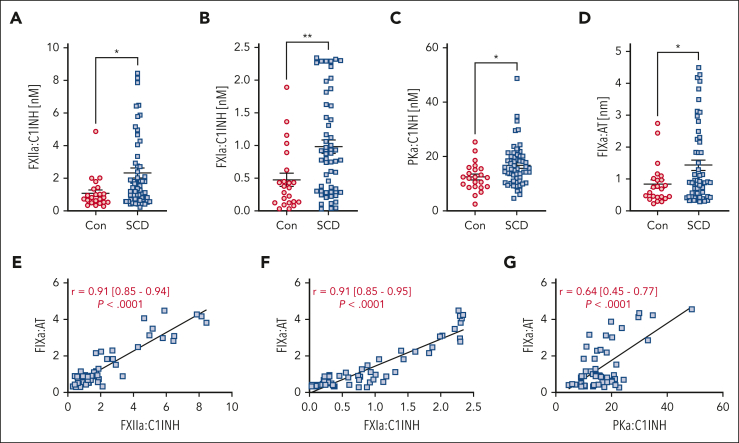
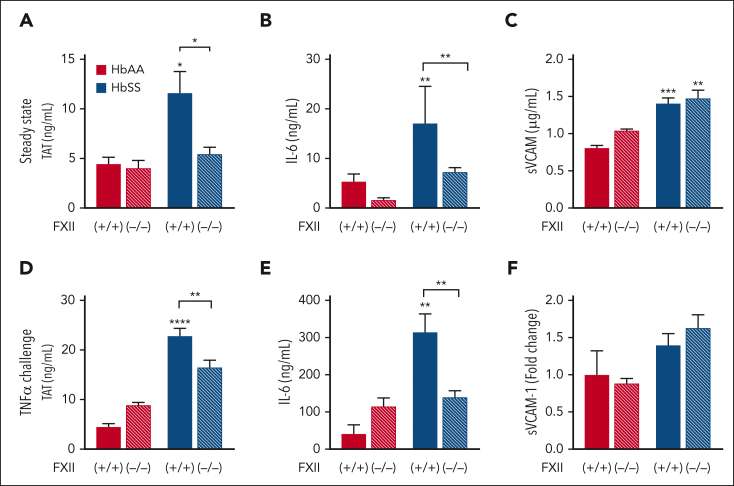
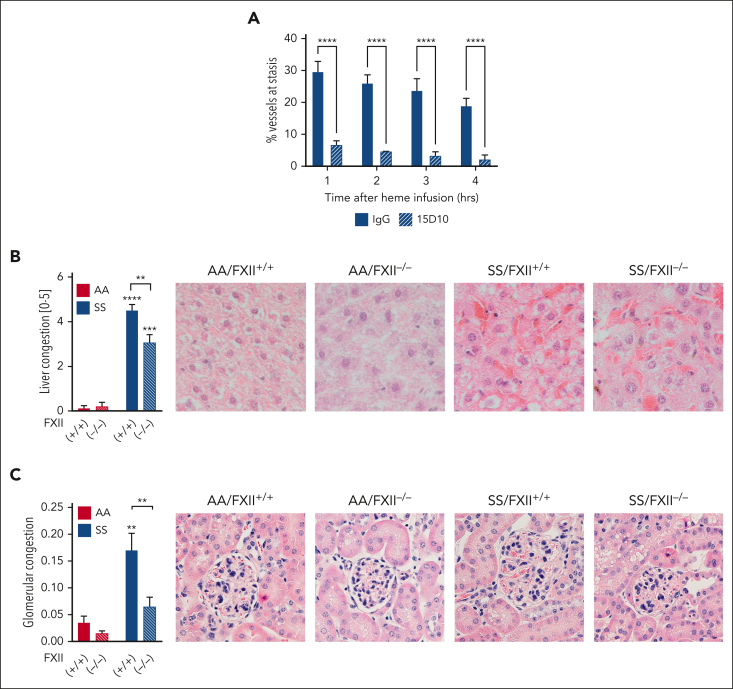
A hypercoagulable state, chronic inflammation, and increased risk of venous thrombosis and stroke are prominent features in patients with sickle cell disease (SCD). Coagulation factor XII (FXII) triggers activation of the contact system that is known to be involved in both thrombosis and inflammation, but not in physiological hemostasis. Therefore, we investigated whether FXII contributes to the prothrombotic and inflammatory complications associated with SCD. We found that when compared with healthy controls, patients with SCD exhibit increased circulating biomarkers of FXII activation that are associated with increased activation of the contact pathway. We also found that FXII, but not tissue factor, contributes to enhanced thrombin generation and systemic inflammation observed in sickle cell mice challenged with tumor necrosis factor α. In addition, FXII inhibition significantly reduced experimental venous thrombosis, congestion, and microvascular stasis in a mouse model of SCD. Moreover, inhibition of FXII attenuated brain damage and reduced neutrophil adhesion to the brain vasculature of sickle cell mice after ischemia/reperfusion induced by transient middle cerebral artery occlusion. Finally, we found higher FXII, urokinase plasminogen activator receptor, and αMβ2 integrin expression in neutrophils of patients with SCD compared with healthy controls. Our data indicate that targeting FXII effectively reduces experimental thromboinflammation and vascular complications in a mouse model of SCD, suggesting that FXII inhibition may provide a safe approach for interference with inflammation, thrombotic complications, and vaso-occlusion in patients with SCD.
Conflict of interest statement
Conflict-of-interest disclosure: R.P. and E.M.S. have received research funding from CSL. A.G. and M.W. are employees of Aronora, Inc. J.D.B. and G.M.V. have received research funding from Omeros, CSL Behring, Hillhurst Biopharmaceuticals, and Astellas/Mitobridge. J.D.B. is a consultant for Astellas/Mitobridge. G.M.V. is a consultant for Sanofi and Astellas/Mitobridge. The remaining authors declare no competing financial interests.
Figures
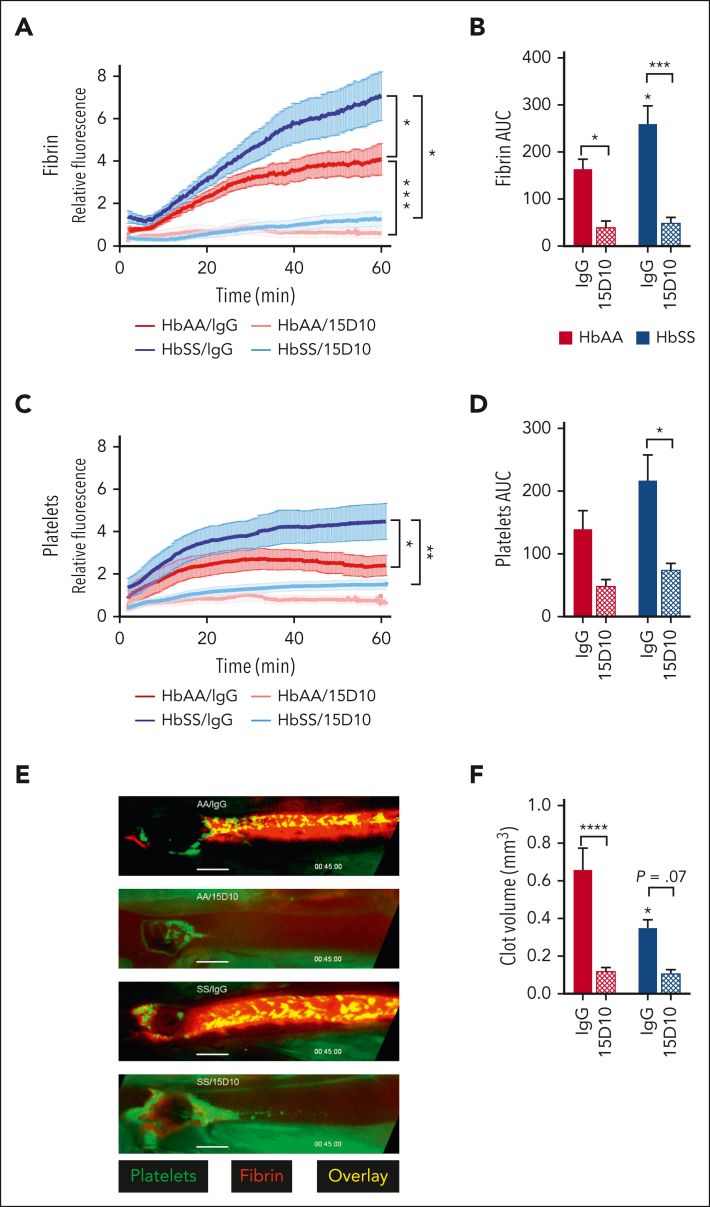
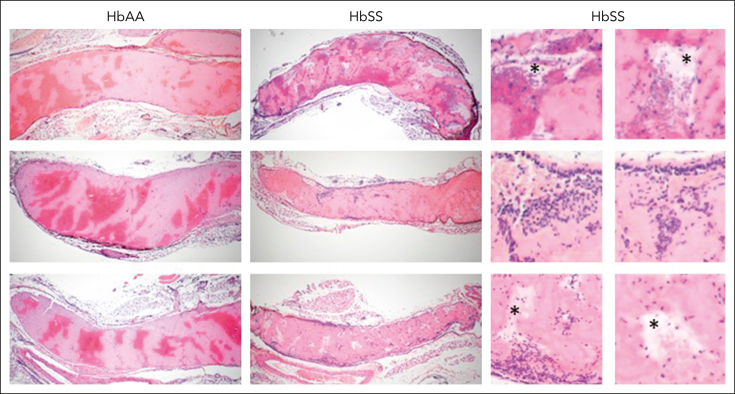
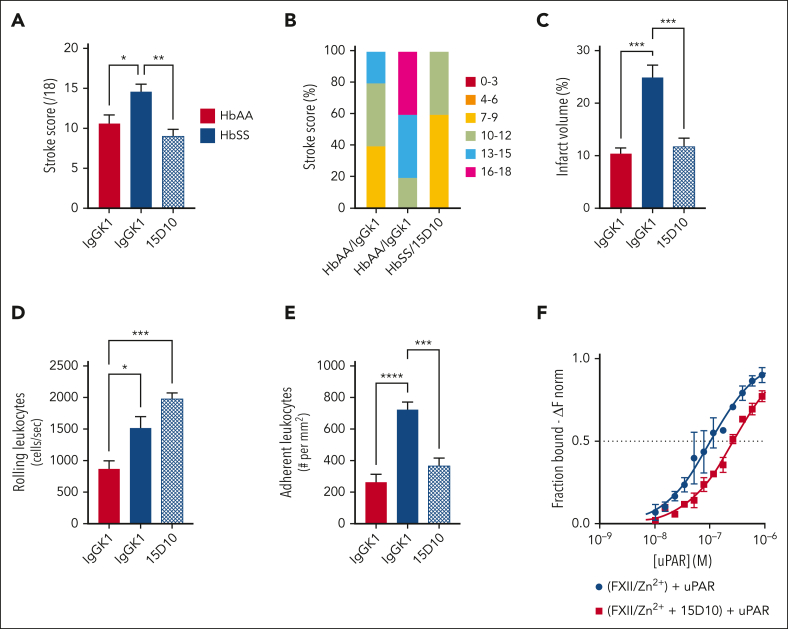
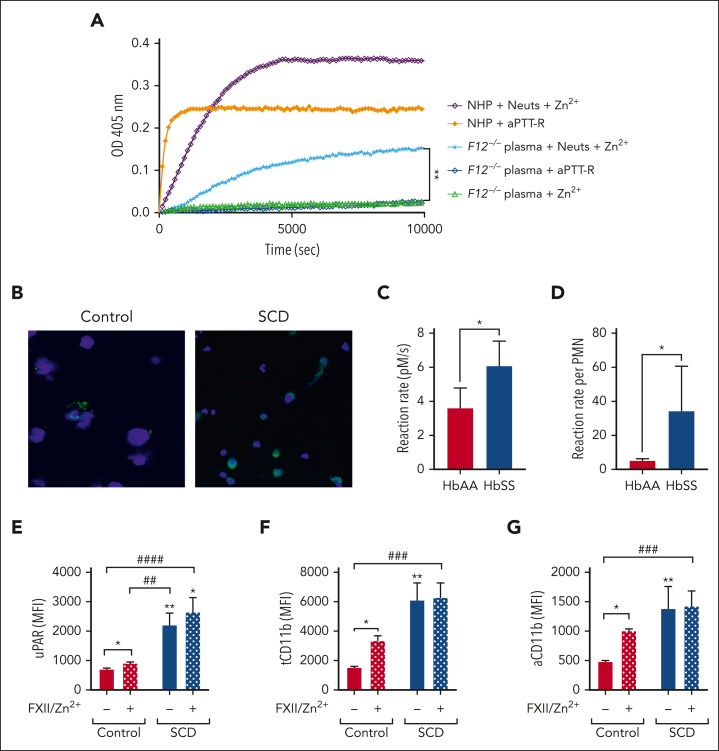
Comment in
-
FXII and sickle cell: the clot thickens.Blood. 2023 Apr 13;141(15):1787-1789. doi: 10.1182/blood.2023019642. Blood. 2023. PMID: 37052945 Free PMC article. No abstract available.
References
-
- Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11(2):129–151. - PubMed
-
- Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. - PubMed
-
- Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: a review. JAMA. 2022;328(1):57–68. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 HL117659/HL/NHLBI NIH HHS/United States
- R44 HL126235/HL/NHLBI NIH HHS/United States
- R01 HL155193/HL/NHLBI NIH HHS/United States
- R01 HL142604/HL/NHLBI NIH HHS/United States
- R00 HL144817/HL/NHLBI NIH HHS/United States
- I01 BX003851/BX/BLRD VA/United States
- T32 HL007149/HL/NHLBI NIH HHS/United States
- R01 HL114567/HL/NHLBI NIH HHS/United States
- R01 HL157441/HL/NHLBI NIH HHS/United States
- R01 HL137695/HL/NHLBI NIH HHS/United States
- R35 HL140025/HL/NHLBI NIH HHS/United States
- K99 HL144817/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
