

Stochastic phenotypes in RAS-dependent developmental diseases
- PMID: 36706752
- PMCID: PMC10026697
- DOI: 10.1016/j.cub.2023.01.008
Stochastic phenotypes in RAS-dependent developmental diseases
Abstract
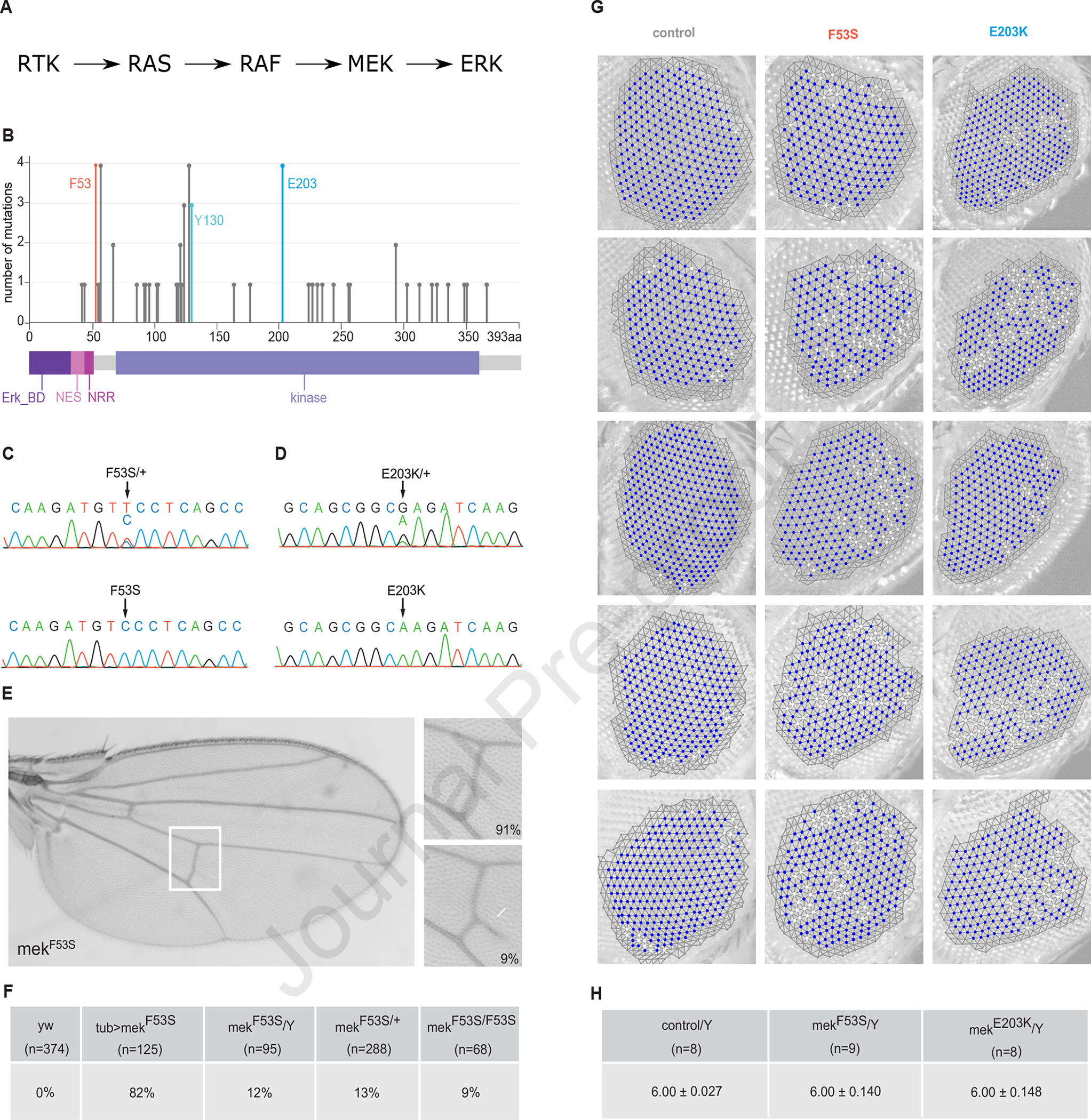
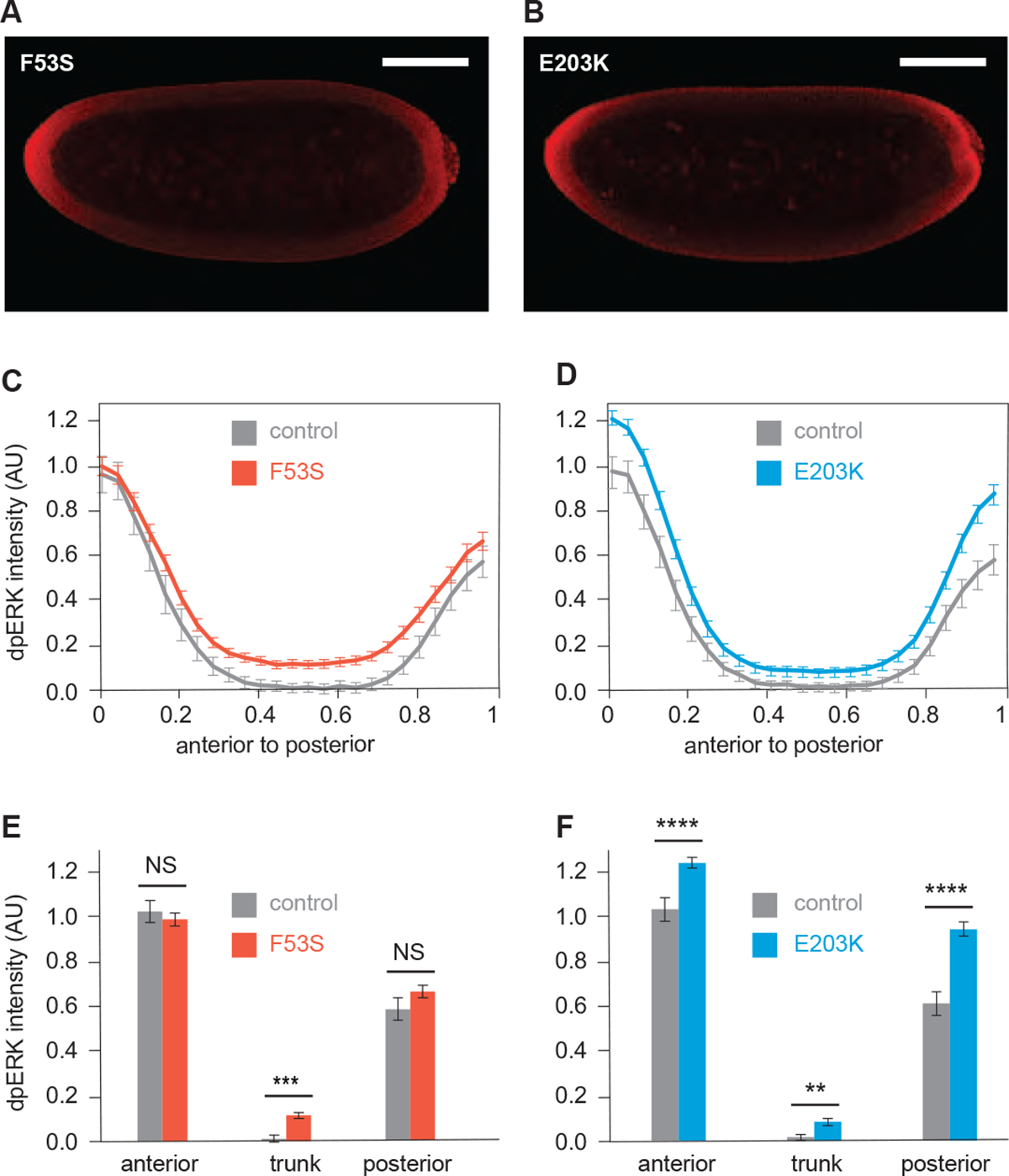
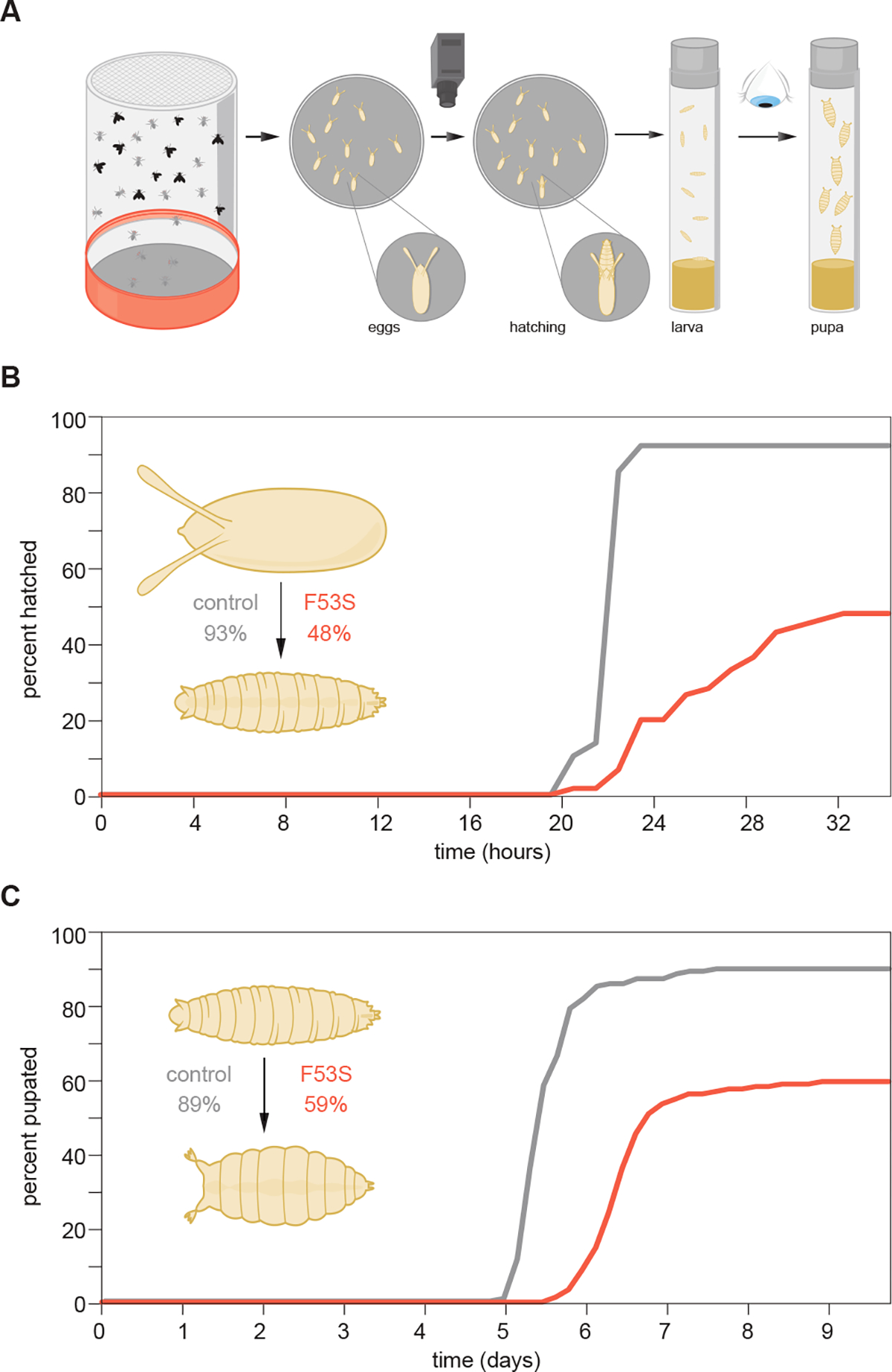
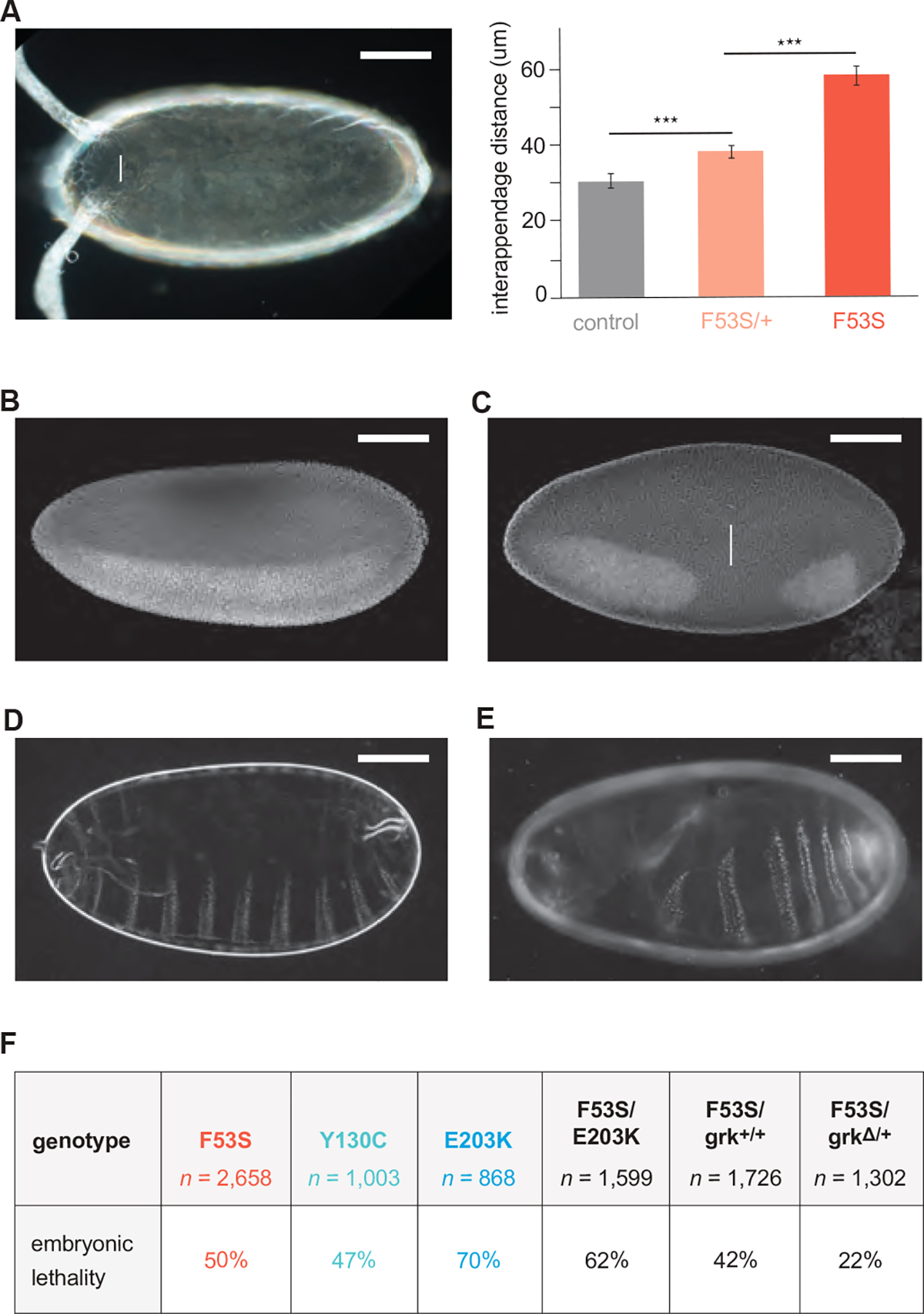
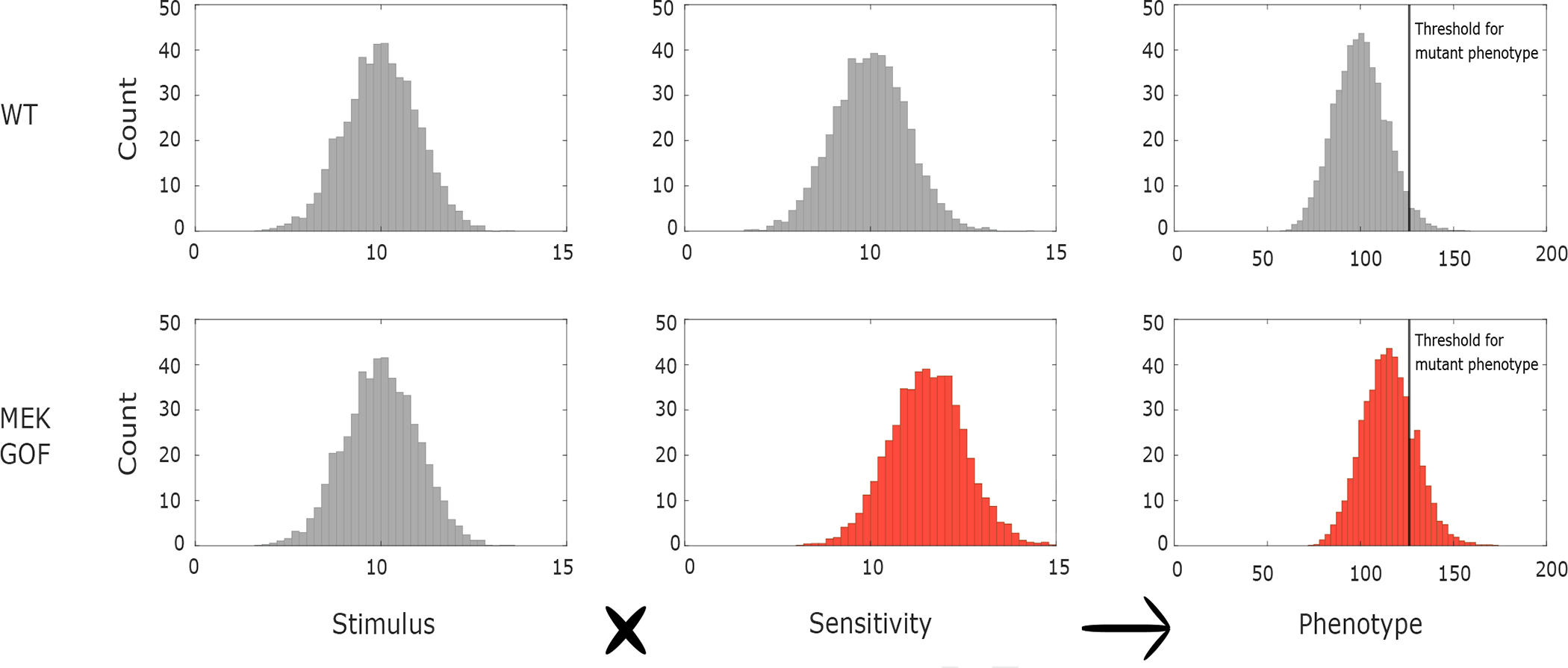
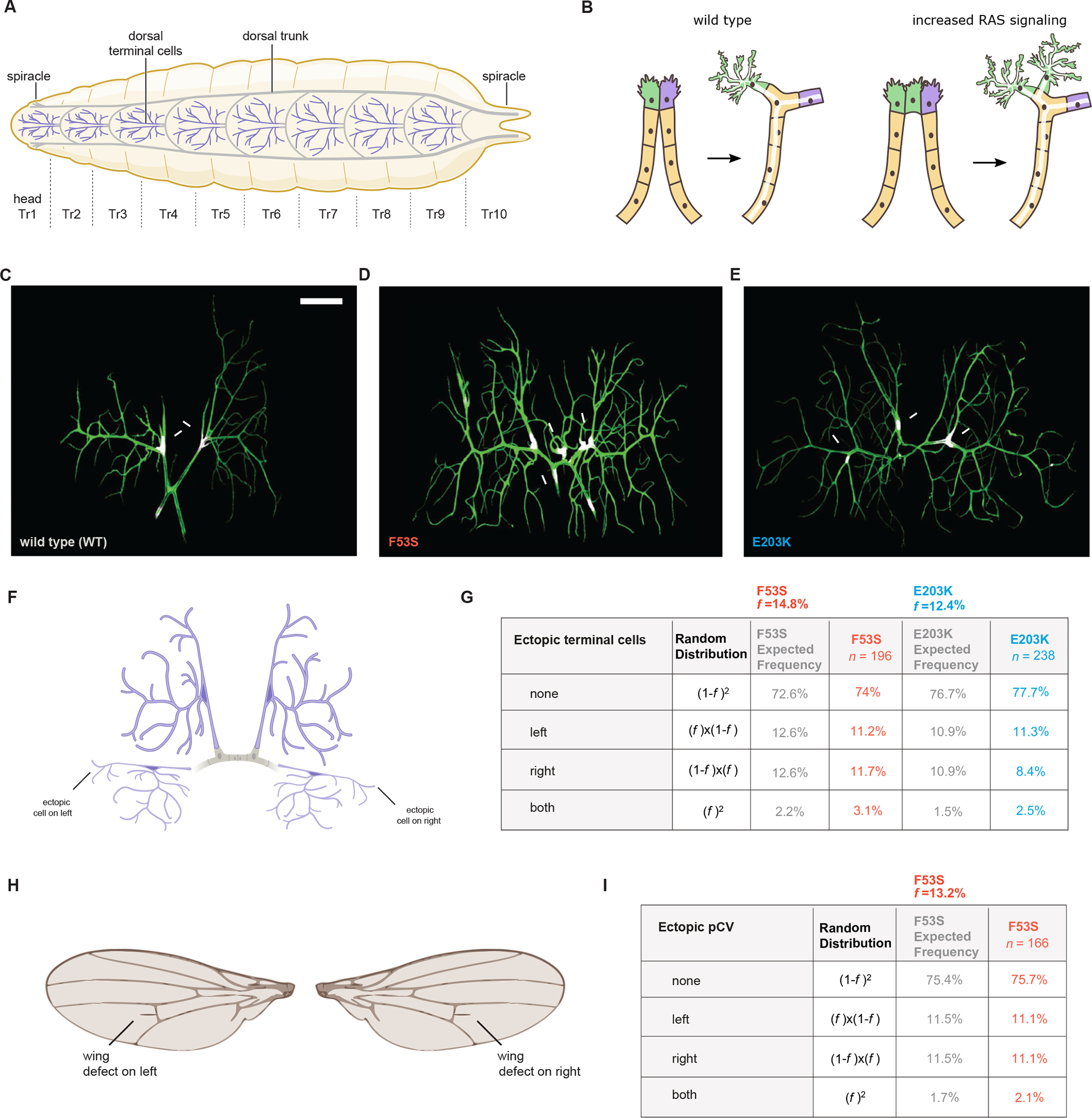
Germline mutations upregulating RAS signaling are associated with multiple developmental disorders. A hallmark of these conditions is that the same mutation may present vastly different phenotypes in different individuals, even in monozygotic twins. Here, we demonstrate how the origins of such largely unexplained phenotypic variations may be dissected using highly controlled studies in Drosophila that have been gene edited to carry activating variants of MEK, a core enzyme in the RAS pathway. This allowed us to measure the small but consistent increase in signaling output of such alleles in vivo. The fraction of mutation carriers reaching adulthood was strongly reduced, but most surviving animals had normal RAS-dependent structures. We rationalize these results using a stochastic signaling model and support it by quantifying cell fate specification errors in bilaterally symmetric larval trachea, a RAS-dependent structure that allows us to isolate the effects of mutations from potential contributions of genetic modifiers and environmental differences. We propose that the small increase in signaling output shifts the distribution of phenotypes into a regime, where stochastic variation causes defects in some individuals, but not in others. Our findings shed light on phenotypic heterogeneity of developmental disorders caused by deregulated RAS signaling and offer a framework for investigating causal effects of other pathogenic alleles and mild mutations in general.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
