

This is a preprint.
ConDoR: Tumor phylogeny inference with a copy-number constrained mutation loss model
- PMID: 36711528
- PMCID: PMC9882003
- DOI: 10.1101/2023.01.05.522408
ConDoR: Tumor phylogeny inference with a copy-number constrained mutation loss model
Update in
-
ConDoR: tumor phylogeny inference with a copy-number constrained mutation loss model.Genome Biol. 2023 Nov 30;24(1):272. doi: 10.1186/s13059-023-03106-5. Genome Biol. 2023. PMID: 38037115 Free PMC article.
Abstract
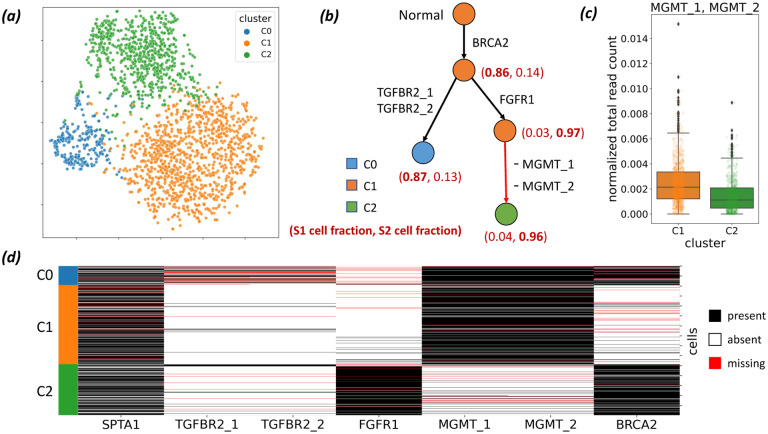
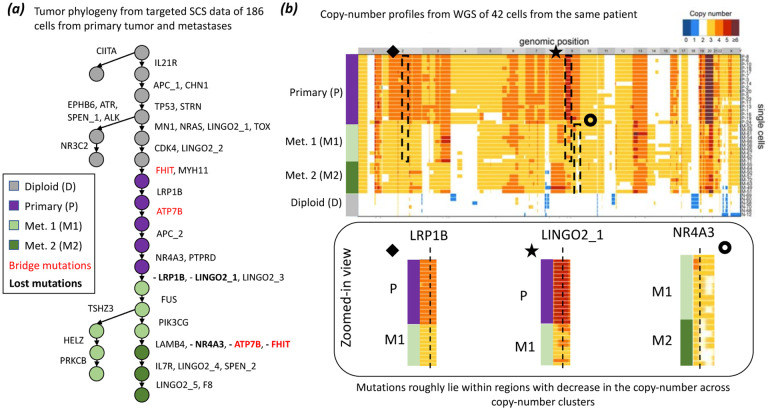
Tumors consist of subpopulations of cells that harbor distinct collections of somatic mutations. These mutations range in scale from single nucleotide variants (SNVs) to large-scale copy-number aberrations (CNAs). While many approaches infer tumor phylogenies using SNVs as phylogenetic markers, CNAs that overlap SNVs may lead to erroneous phylogenetic inference. Specifically, an SNV may be lost in a cell due to a deletion of the genomic segment containing the SNV. Unfortunately, no current single-cell DNA sequencing (scDNA-seq) technology produces accurate measurements of both SNVs and CNAs. For instance, recent targeted scDNA-seq technologies, such as Mission Bio Tapestri, measure SNVs with high fidelity in individual cells, but yield much less reliable measurements of CNAs. We introduce a new evolutionary model, the constrained k-Dollo model, that uses SNVs as phylogenetic markers and partial information about CNAs in the form of clustering of cells with similar copy-number profiles. This copy-number clustering constrains where loss of SNVs can occur in the phylogeny. We develop ConDoR (Constrained Dollo Reconstruction), an algorithm to infer tumor phylogenies from targeted scDNA-seq data using the constrained k-Dollo model. We show that ConDoR outperforms existing methods on simulated data. We use ConDoR to analyze a new multi-region targeted scDNA-seq dataset of 2153 cells from a pancreatic ductal adenocarcinoma (PDAC) tumor and produce a more plausible phylogeny compared to existing methods that conforms to histological results for the tumor from a previous study. We also analyze a metastatic colorectal cancer dataset, deriving a more parsimonious phylogeny than previously published analyses and with a simpler monoclonal origin of metastasis compared to the original study.
Code availability: Software is available at https://github.com/raphael-group/constrained-Dollo.
Conflict of interest statement
Competing interests The authors declare that they have no competing interests.
Figures


References
-
- Tabassum Doris P and Polyak Kornelia. Tumorigenesis: it takes a village. Nature Reviews Cancer, 15(8): 473–483, 2015. - PubMed
-
- Amirouchene-Angelozzi Nabil, Swanton Charles, and Bardelli Alberto. Tumor evolution as a therapeutic target the impact of tumor evolution in precision medicine. Cancer discovery, 7(8):805–817, 2017. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources