

This is a preprint.
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation
- PMID: 36711673
- PMCID: PMC9882142
- DOI: 10.1101/2023.01.12.523790
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation
Update in
-
Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation.Nat Methods. 2023 Oct;20(10):1483-1492. doi: 10.1038/s41592-023-01993-x. Epub 2023 Sep 14. Nat Methods. 2023. PMID: 37710018 Free PMC article.
Abstract
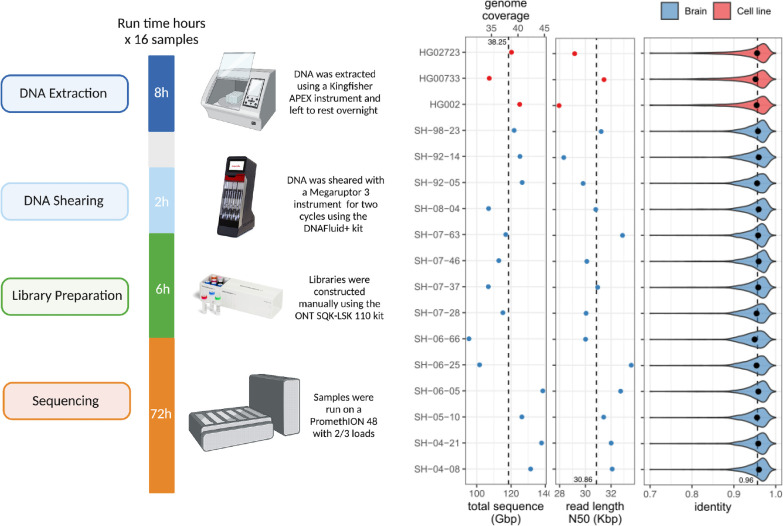
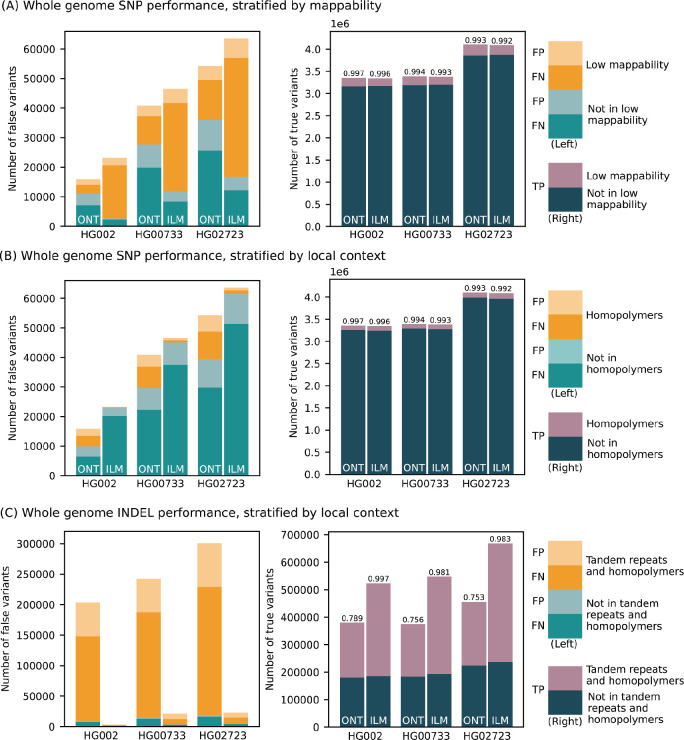
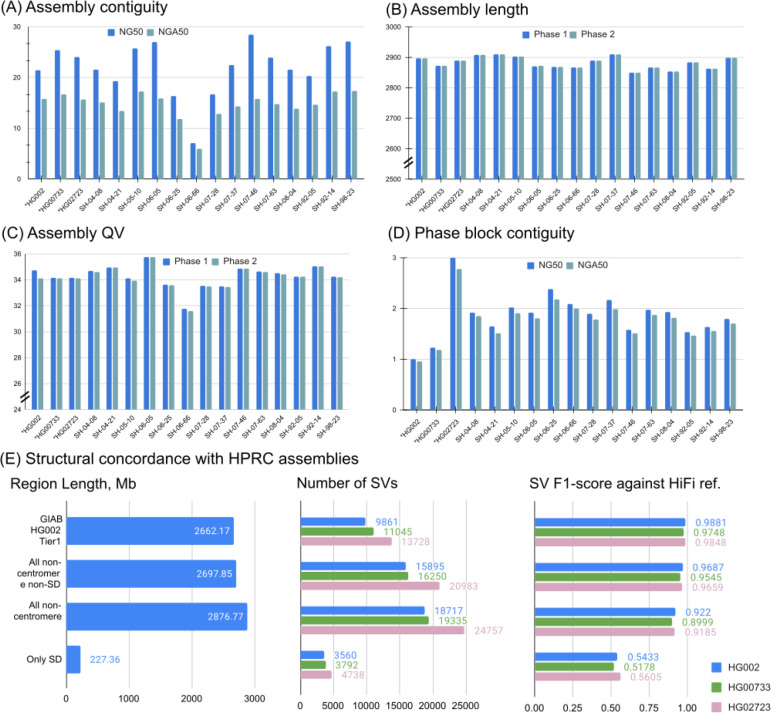
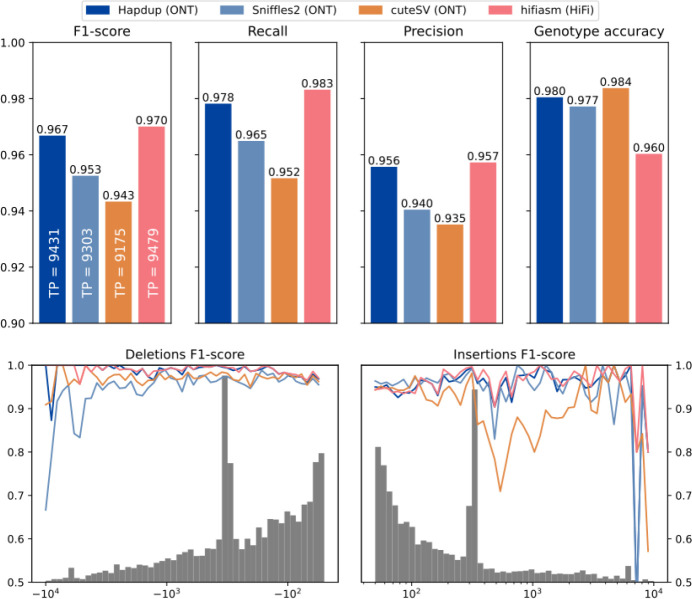
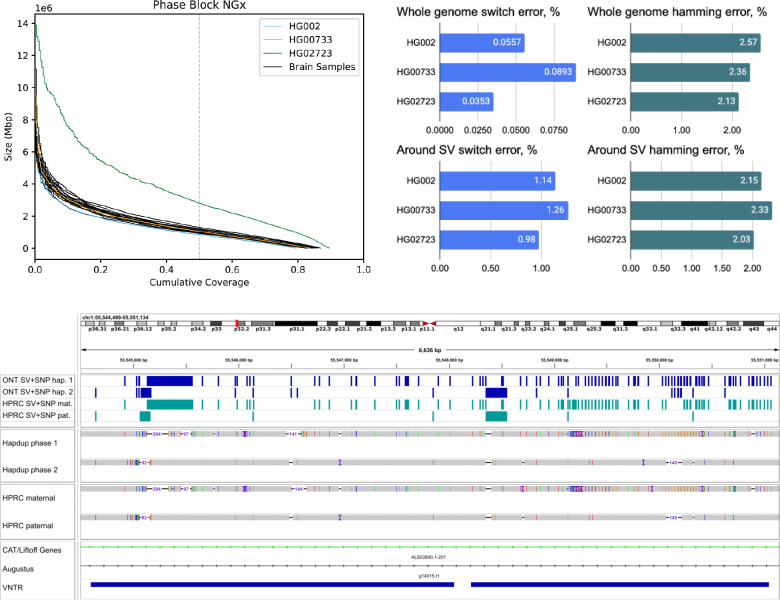
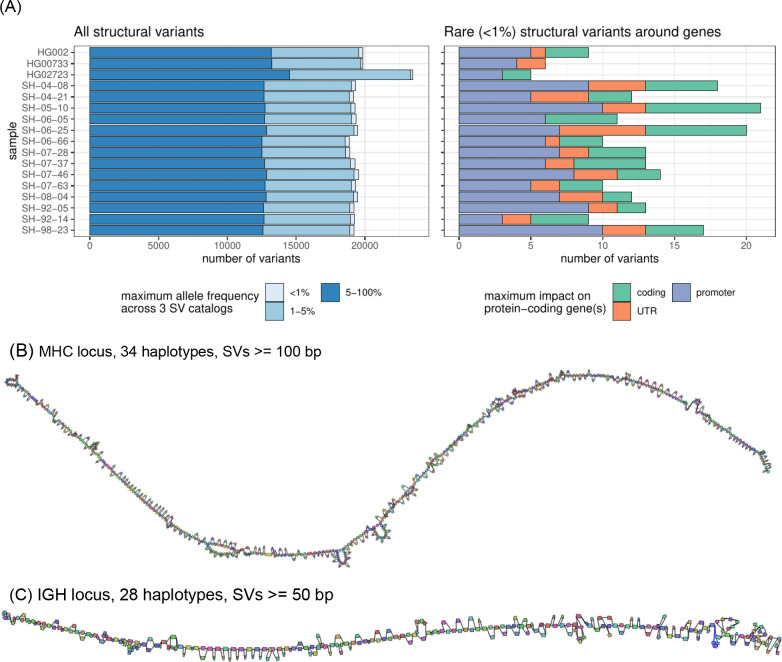
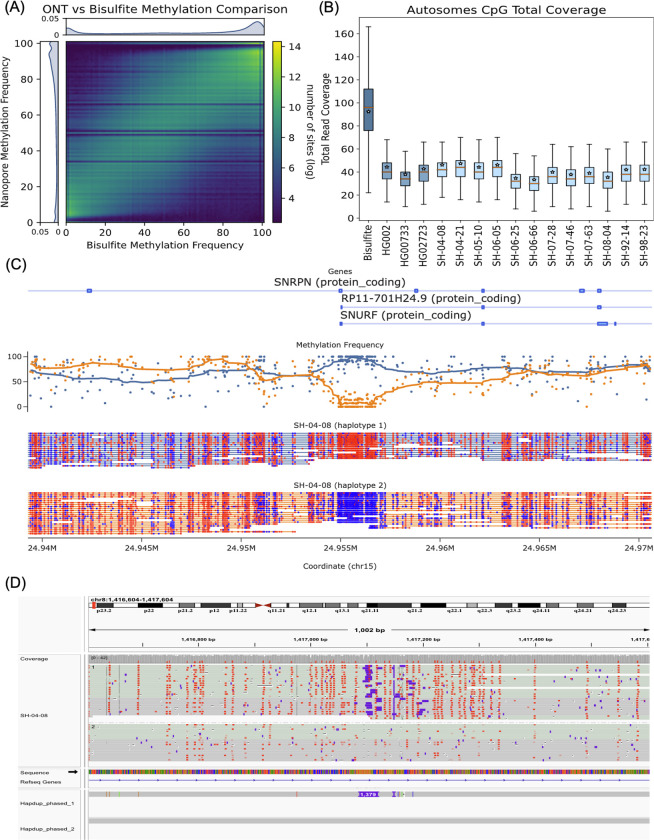
Long-read sequencing technologies substantially overcome the limitations of short-reads but to date have not been considered as feasible replacement at scale due to a combination of being too expensive, not scalable enough, or too error-prone. Here, we develop an efficient and scalable wet lab and computational protocol for Oxford Nanopore Technologies (ONT) long-read sequencing that seeks to provide a genuine alternative to short-reads for large-scale genomics projects. We applied our protocol to cell lines and brain tissue samples as part of a pilot project for the NIH Center for Alzheimer's and Related Dementias (CARD). Using a single PromethION flow cell, we can detect SNPs with F1-score better than Illumina short-read sequencing. Small indel calling remains to be difficult inside homopolymers and tandem repeats, but is comparable to Illumina calls elsewhere. Further, we can discover structural variants with F1-score comparable to state-of the-art methods involving Pacific Biosciences HiFi sequencing and trio information (but at a lower cost and greater throughput). Using ONT based phasing, we can then combine and phase small and structural variants at megabase scales. Our protocol also produces highly accurate, haplotype-specific methylation calls. Overall, this makes large-scale long-read sequencing projects feasible; the protocol is currently being used to sequence thousands of brain-based genomes as a part of the NIH CARD initiative. We provide the protocol and software as open-source integrated pipelines for generating phased variant calls and assemblies.
Conflict of interest statement
Figures
References
-
- 100,000 Genomes Project Pilot Investigators, Smedley D., Smith K. R., Martin A., Thomas E. A., McDonagh E. M., Cipriani V., Ellingford J. M., Arno G., Tucci A., Vandrovcova J., Chan G., Williams H. J., Ratnaike T., Wei W., Stirrups K., Ibanez K., Moutsianas L., Wielscher M., … Caulfield M. (2021). 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. The New England Journal of Medicine, 385(20), 1868–1880. - PMC - PubMed
-
- Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A. J., Kruglyak S., & Saunders C. T. (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics , 32(8), 1220–1222. - PubMed
Publication types
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- T32 HG012344/HG/NHGRI NIH HHS/United States
- P01 AG000538/AG/NIA NIH HHS/United States
- P30 AG072980/AG/NIA NIH HHS/United States
- OT3 HL142481/HL/NHLBI NIH HHS/United States
- OT2 OD033761/OD/NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24 HG010262/HG/NHGRI NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- P30 AG019610/AG/NIA NIH HHS/United States
- ZIA AG000538/ImNIH/Intramural NIH HHS/United States
- R01 HG009190/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- ZIA NS003154/ImNIH/Intramural NIH HHS/United States
- U24 NS072026/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous