

This is a preprint.
Ferroptosis Integrates Mitochondrial Derangements and Pathological Inflammation to Promote Pulmonary Hypertension
- PMID: 36712076
- PMCID: PMC9882268
- DOI: 10.1101/2023.01.19.524721
Ferroptosis Integrates Mitochondrial Derangements and Pathological Inflammation to Promote Pulmonary Hypertension
Abstract
Background: Mitochondrial dysfunction, characterized by impaired lipid metabolism and heightened reactive oxygen species (ROS) generation, results in lipid peroxidation and ferroptosis. Ferroptosis is an inflammatory mode of cell death that promotes complement activation and macrophage recruitment. In pulmonary arterial hypertension (PAH), pulmonary arterial endothelial cells (PAEC) exhibit cellular phenotypes that promote ferroptosis. Moreover, there is ectopic complement deposition and inflammatory macrophage accumulation in the pulmonary vasculature. However, the effects of ferroptosis inhibition on these pathogenic mechanisms and the cellular landscape of the pulmonary vasculature are incompletely defined.
Methods: Multi-omics and physiological analyses evaluated how ferroptosis inhibition modulated preclinical PAH. The impact of AAV1-mediated expression of the pro-ferroptotic protein ACSL4 on PAH was determined, and a genetic association study in humans further probed the relationship between ferroptosis and pulmonary hypertension (PH).
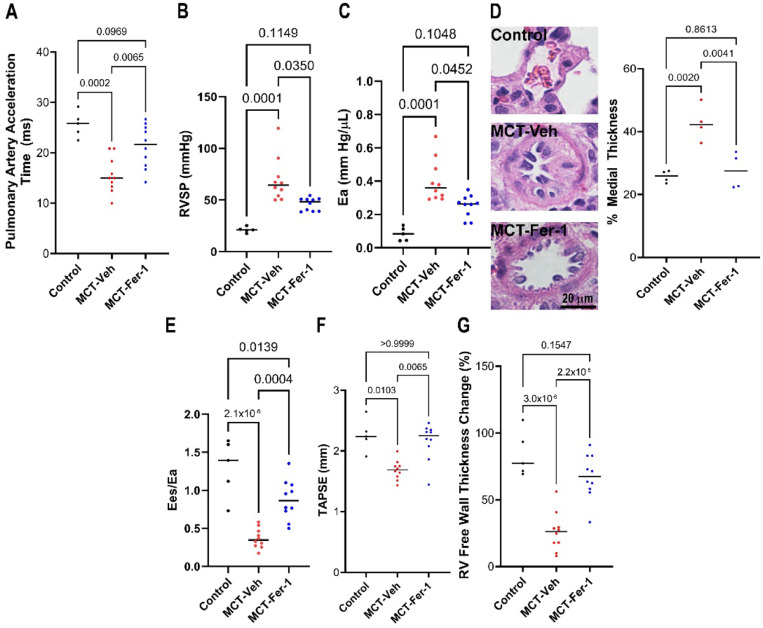
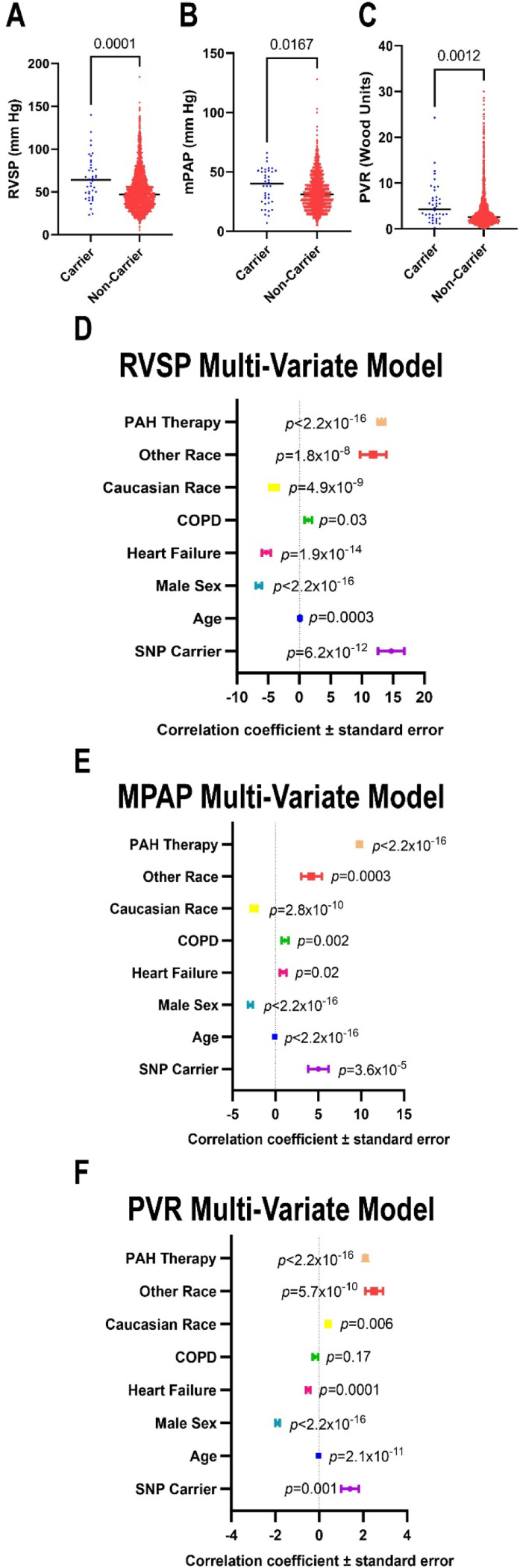
Results: Ferrostatin-1, a small-molecule ferroptosis inhibitor, mitigated PAH severity in monocrotaline rats. RNA-seq and proteomics analyses demonstrated ferroptosis was associated with PAH severity. RNA-seq, proteomics, and confocal microscopy revealed complement activation and pro-inflammatory cytokines/chemokines were suppressed by ferrostatin-1. Additionally, ferrostatin-1 combatted changes in endothelial, smooth muscle, and interstitial macrophage abundance and gene activation patterns as revealed by deconvolution RNA-seq. Ferroptotic PAEC damage associated molecular patterns restructured the transcriptomic signature, mitochondrial morphology, and promoted proliferation of pulmonary artery smooth muscle cells, and created a pro-inflammatory phenotype in monocytes in vitro. AAV1-Acsl4 induced an inflammatory PAH phenotype in rats. Finally, single-nucleotide polymorphisms in six ferroptosis genes identified a potential link between ferroptosis and PH severity in the Vanderbilt BioVU repository.
Conclusions: Ferroptosis promotes PAH through metabolic and inflammatory mechanisms in the pulmonary vasculature.
Figures






Similar articles
-
Ferroptosis-Mediated Inflammation Promotes Pulmonary Hypertension.Circ Res. 2024 Nov 8;135(11):1067-1083. doi: 10.1161/CIRCRESAHA.123.324138. Epub 2024 Oct 18. Circ Res. 2024. PMID: 39421926
-
M1 Macrophage-Derived TNF-α Promotes Pancreatic Cancer Ferroptosis Via p38 MAPK-ACSL4 Pathway.Curr Mol Med. 2025 Jul 10. doi: 10.2174/0115665240374551250630075409. Online ahead of print. Curr Mol Med. 2025. PMID: 40653839
-
Perillaldehyde synergizes with ferroptosis inducers to promote ferroptotic cell death in gastric cancer.Front Cell Dev Biol. 2025 Jun 3;13:1598520. doi: 10.3389/fcell.2025.1598520. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40530331 Free PMC article.
-
Guanylate cyclase stimulators for pulmonary hypertension.Cochrane Database Syst Rev. 2016 Aug 2;2016(8):CD011205. doi: 10.1002/14651858.CD011205.pub2. Cochrane Database Syst Rev. 2016. PMID: 27482837 Free PMC article.
-
Exploring the Economic Burden of Pulmonary Arterial Hypertension and Its Relation to Disease Severity and Treatment Escalation: A Systematic Literature Review.Pharmacoeconomics. 2025 Jul;43(7):741-760. doi: 10.1007/s40273-025-01492-1. Epub 2025 Apr 17. Pharmacoeconomics. 2025. PMID: 40244370 Free PMC article.
References
Publication types
Grants and funding
- R01 DK091538/DK/NIDDK NIH HHS/United States
- R01 HL162927/HL/NHLBI NIH HHS/United States
- R01 FD007627/FD/FDA HHS/United States
- T32 HL144472/HL/NHLBI NIH HHS/United States
- K08 HL168166/HL/NHLBI NIH HHS/United States
- R01 HL166843/HL/NHLBI NIH HHS/United States
- R61 HL158941/HL/NHLBI NIH HHS/United States
- R01 AI165553/AI/NIAID NIH HHS/United States
- R01 HL155278/HL/NHLBI NIH HHS/United States
- R01 HL158795/HL/NHLBI NIH HHS/United States
- T32 HL072742/HL/NHLBI NIH HHS/United States
- R01 HL146588/HL/NHLBI NIH HHS/United States
- R01 HL163960/HL/NHLBI NIH HHS/United States
- R01 DK124845/DK/NIDDK NIH HHS/United States
- K08 HL140100/HL/NHLBI NIH HHS/United States
- S10 OD028717/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources