

LKB1-Dependent Regulation of TPI1 Creates a Divergent Metabolic Liability between Human and Mouse Lung Adenocarcinoma
- PMID: 36715544
- PMCID: PMC10068449
- DOI: 10.1158/2159-8290.CD-22-0805
LKB1-Dependent Regulation of TPI1 Creates a Divergent Metabolic Liability between Human and Mouse Lung Adenocarcinoma
Abstract
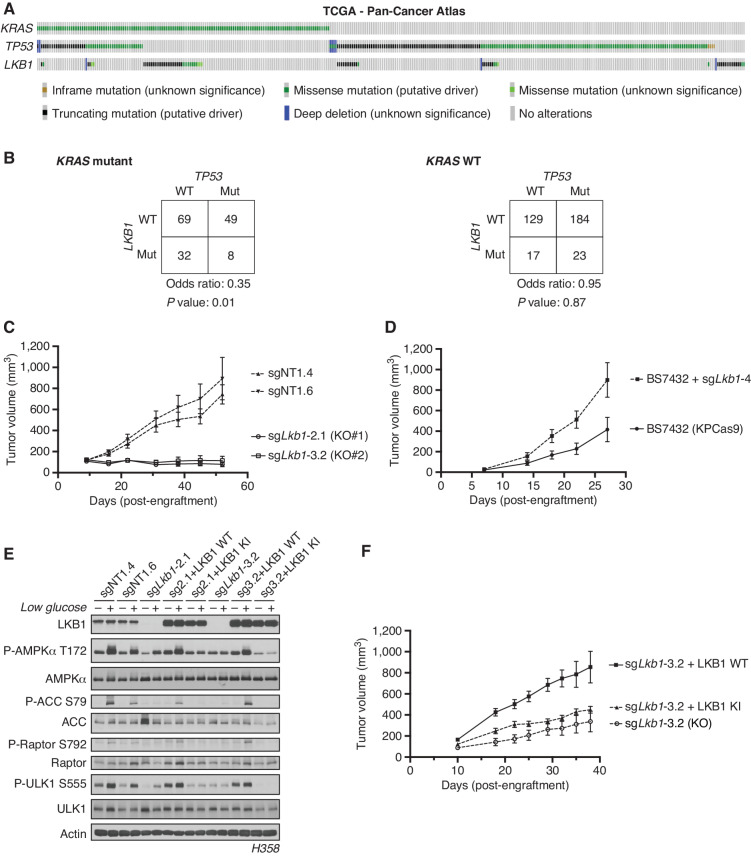
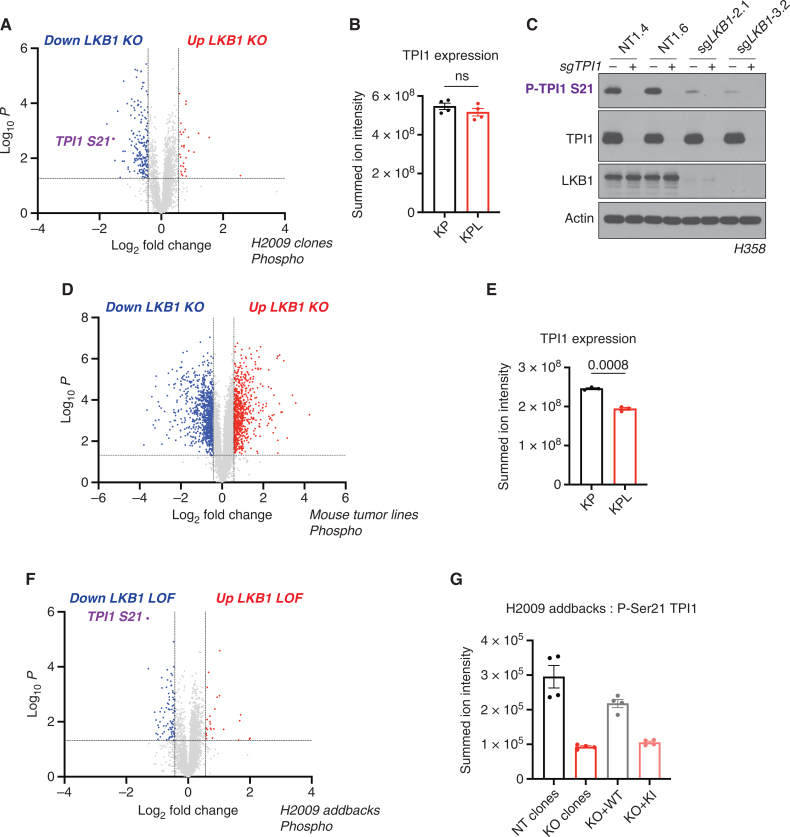
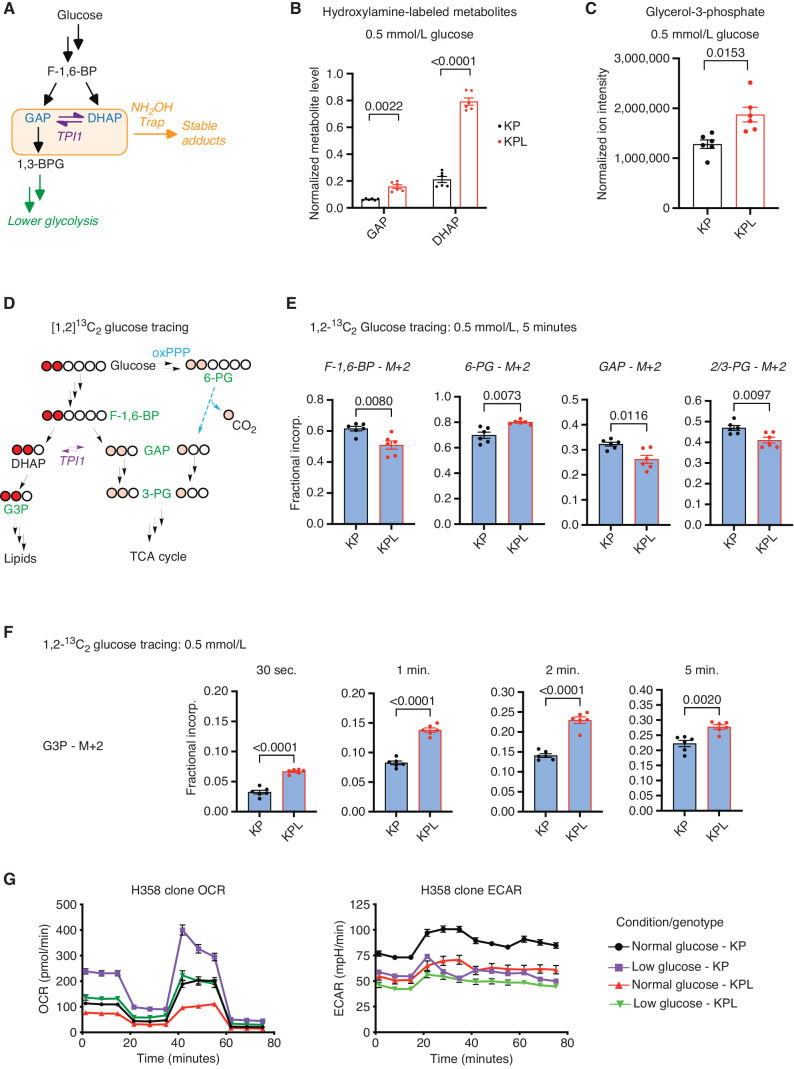
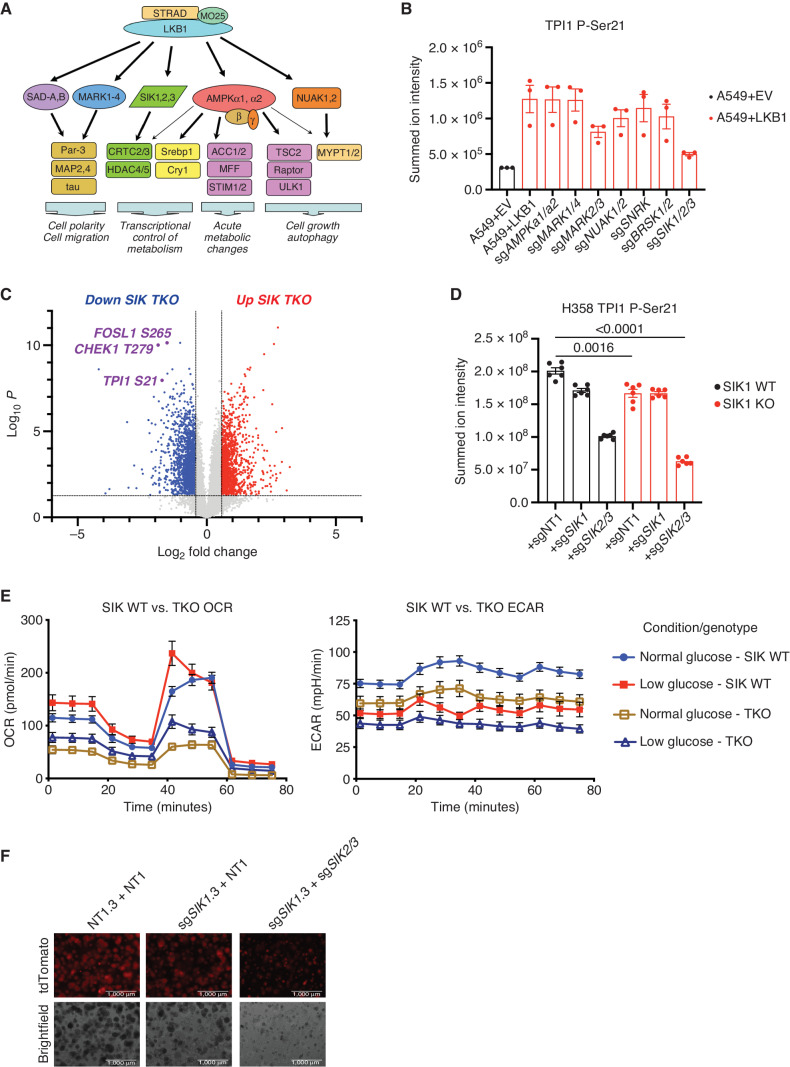
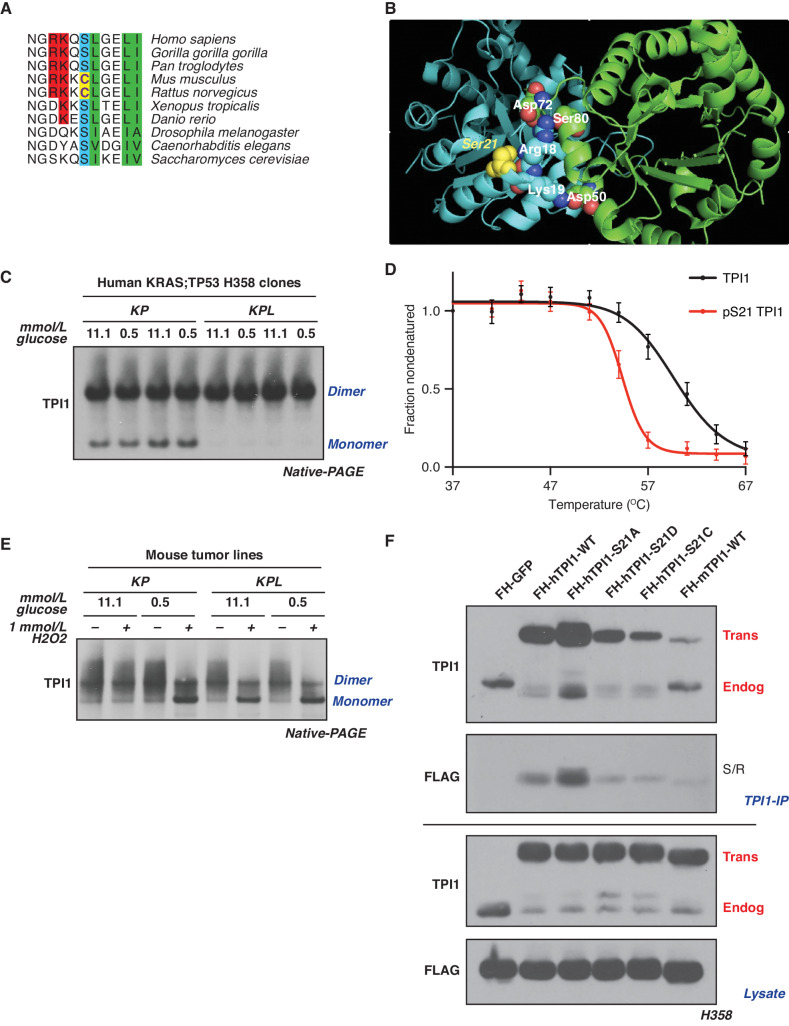
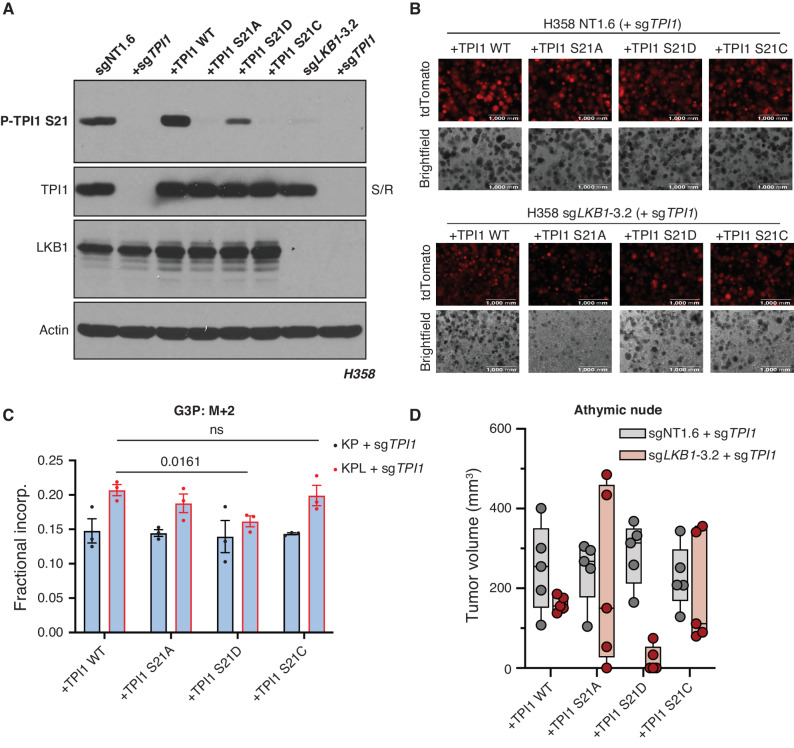
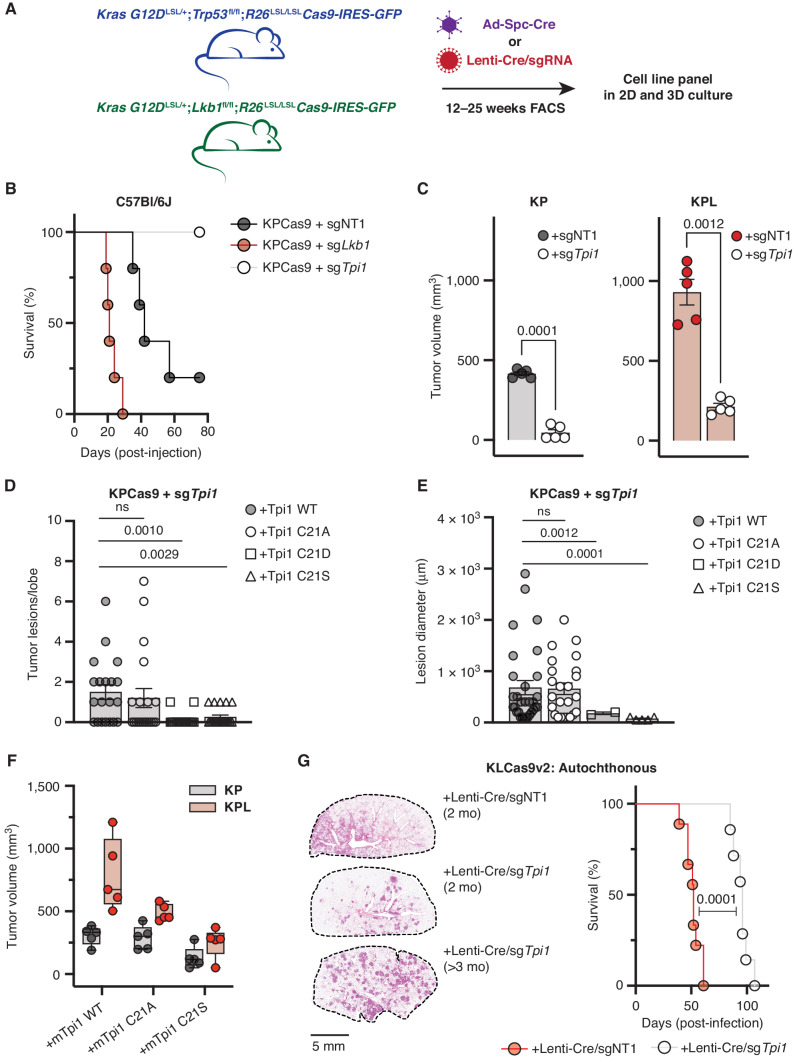
KRAS is the most frequently mutated oncogene in human lung adenocarcinomas (hLUAD), and activating mutations frequently co-occur with loss-of-function mutations in TP53 or STK11/LKB1. However, mutation of all three genes is rarely observed in hLUAD, even though engineered comutation is highly aggressive in mouse lung adenocarcinoma (mLUAD). Here, we provide a mechanistic explanation for this difference by uncovering an evolutionary divergence in the regulation of triosephosphate isomerase (TPI1). In hLUAD, TPI1 activity is regulated via phosphorylation at Ser21 by the salt inducible kinases (SIK) in an LKB1-dependent manner, modulating flux between the completion of glycolysis and production of glycerol lipids. In mice, Ser21 of TPI1 is a Cys residue that can be oxidized to alter TPI1 activity without a need for SIKs or LKB1. Our findings suggest this metabolic flexibility is critical in rapidly growing cells with KRAS and TP53 mutations, explaining why the loss of LKB1 creates a liability in these tumors.
Significance: Utilizing phosphoproteomics and metabolomics in genetically engineered human cell lines and genetically engineered mouse models (GEMM), we uncover an evolutionary divergence in metabolic regulation within a clinically relevant genotype of human LUAD with therapeutic implications. Our data provide a cautionary example of the limits of GEMMs as tools to study human diseases such as cancers. This article is highlighted in the In This Issue feature, p. 799.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
- 2159-8274. doi: 10.1158/2159-8290.CD-13-4-ITI
References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
