

SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia
- PMID: 36718090
- PMCID: PMC10319774
- DOI: 10.1093/brain/awac460
SPTSSA variants alter sphingolipid synthesis and cause a complex hereditary spastic paraplegia
Abstract
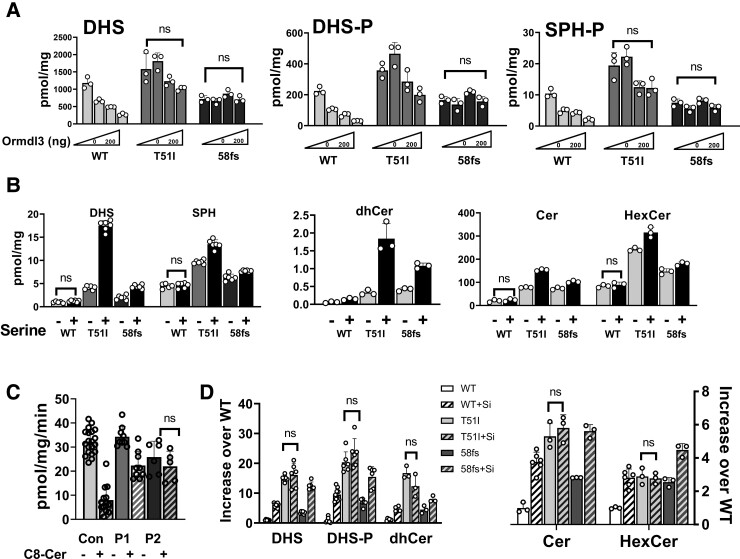
Sphingolipids are a diverse family of lipids with critical structural and signalling functions in the mammalian nervous system, where they are abundant in myelin membranes. Serine palmitoyltransferase, the enzyme that catalyses the rate-limiting reaction of sphingolipid synthesis, is composed of multiple subunits including an activating subunit, SPTSSA. Sphingolipids are both essential and cytotoxic and their synthesis must therefore be tightly regulated. Key to the homeostatic regulation are the ORMDL proteins that are bound to serine palmitoyltransferase and mediate feedback inhibition of enzymatic activity when sphingolipid levels become excessive. Exome sequencing identified potential disease-causing variants in SPTSSA in three children presenting with a complex form of hereditary spastic paraplegia. The effect of these variants on the catalytic activity and homeostatic regulation of serine palmitoyltransferase was investigated in human embryonic kidney cells, patient fibroblasts and Drosophila. Our results showed that two different pathogenic variants in SPTSSA caused a hereditary spastic paraplegia resulting in progressive motor disturbance with variable sensorineural hearing loss and language/cognitive dysfunction in three individuals. The variants in SPTSSA impaired the negative regulation of serine palmitoyltransferase by ORMDLs leading to excessive sphingolipid synthesis based on biochemical studies and in vivo studies in Drosophila. These findings support the pathogenicity of the SPTSSA variants and point to excessive sphingolipid synthesis due to impaired homeostatic regulation of serine palmitoyltransferase as responsible for defects in early brain development and function.
Keywords: SPTSSA; ORMDLs; hereditary spastic paraplegia; serine palmitoyltransferase; sphingolipids.
Published by Oxford University Press on behalf of the Guarantors of Brain 2023.
Conflict of interest statement
B.P.K. is an inventor on patents and patent applications filed by Mass General Brigham that describe genome engineering technologies. B.P.K. consults for Avectas Inc., EcoR1 capital and ElevateBio, and is an advisor to Acrigen Biosciences, Life Edit Therapeutics and Prime Medicines. S.S. has received consulting fees from GLG, Guidepoint (which connected to a client, Fortress Biotech), Novartis, ExpertConnect and Orchard Therapeutics. F.E. has received consulting fees for UptoDate, Prime Medical Education, bluebird bio, Takeda and SwanBio Therapeutics. All other authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
