

Neuron-oligodendrocyte potassium shuttling at nodes of Ranvier protects against inflammatory demyelination
- PMID: 36719741
- PMCID: PMC10065072
- DOI: 10.1172/JCI164223
Neuron-oligodendrocyte potassium shuttling at nodes of Ranvier protects against inflammatory demyelination
Abstract
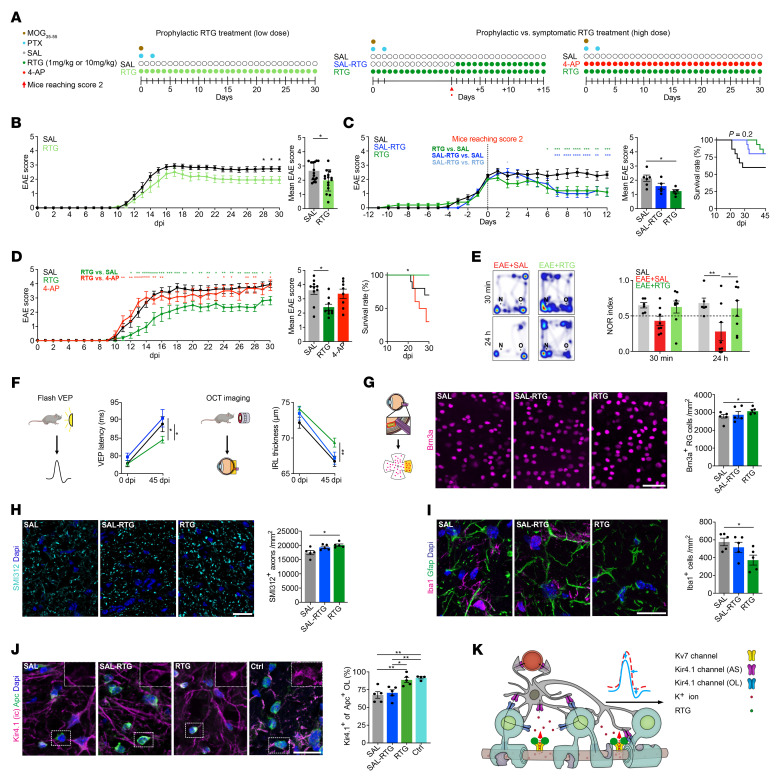
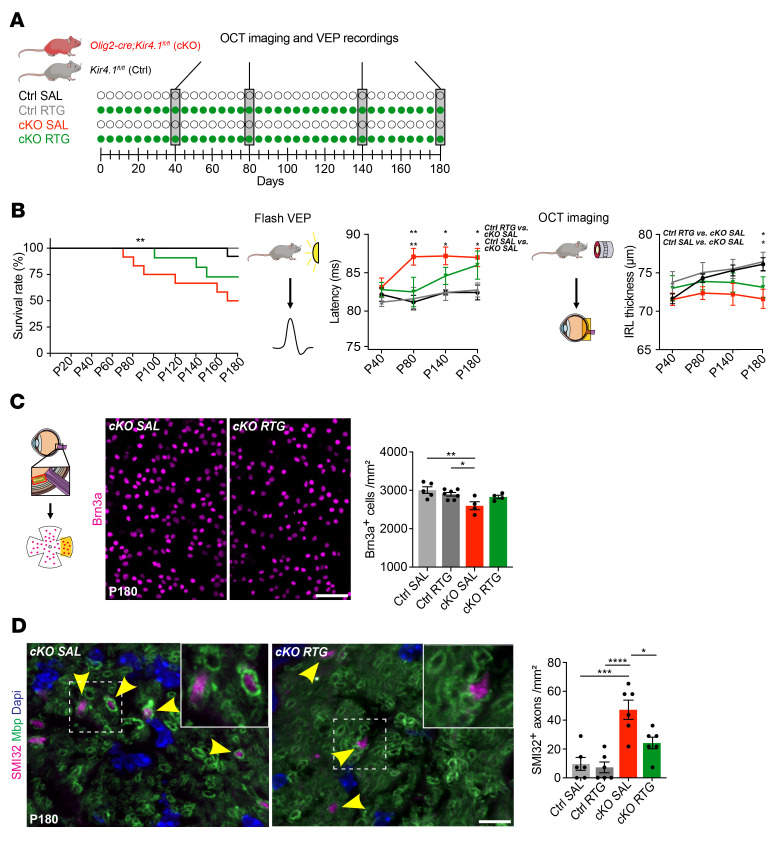
Multiple sclerosis (MS) is a progressive inflammatory demyelinating disease of the CNS. Increasing evidence suggests that vulnerable neurons in MS exhibit fatal metabolic exhaustion over time, a phenomenon hypothesized to be caused by chronic hyperexcitability. Axonal Kv7 (outward-rectifying) and oligodendroglial Kir4.1 (inward-rectifying) potassium channels have important roles in regulating neuronal excitability at and around the nodes of Ranvier. Here, we studied the spatial and functional relationship between neuronal Kv7 and oligodendroglial Kir4.1 channels and assessed the transcriptional and functional signatures of cortical and retinal projection neurons under physiological and inflammatory demyelinating conditions. We found that both channels became dysregulated in MS and experimental autoimmune encephalomyelitis (EAE), with Kir4.1 channels being chronically downregulated and Kv7 channel subunits being transiently upregulated during inflammatory demyelination. Further, we observed that pharmacological Kv7 channel opening with retigabine reduced neuronal hyperexcitability in human and EAE neurons, improved clinical EAE signs, and rescued neuronal pathology in oligodendrocyte-Kir4.1-deficient (OL-Kir4.1-deficient) mice. In summary, our findings indicate that neuron-OL compensatory interactions promoted resilience through Kv7 and Kir4.1 channels and identify pharmacological activation of nodal Kv7 channels as a neuroprotective strategy against inflammatory demyelination.
Keywords: Inflammation; Multiple sclerosis; Neurodegeneration; Neuroscience; Potassium channels.
Figures
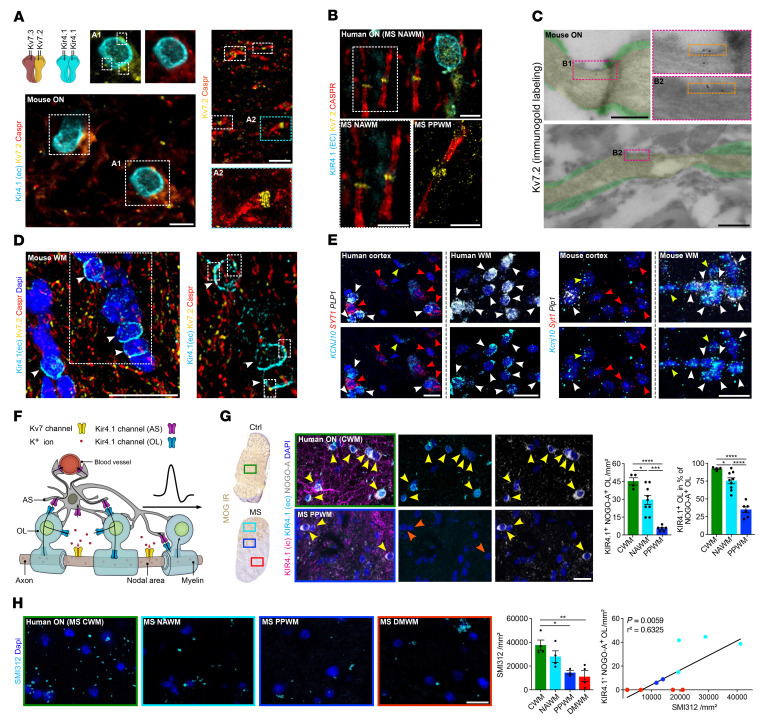
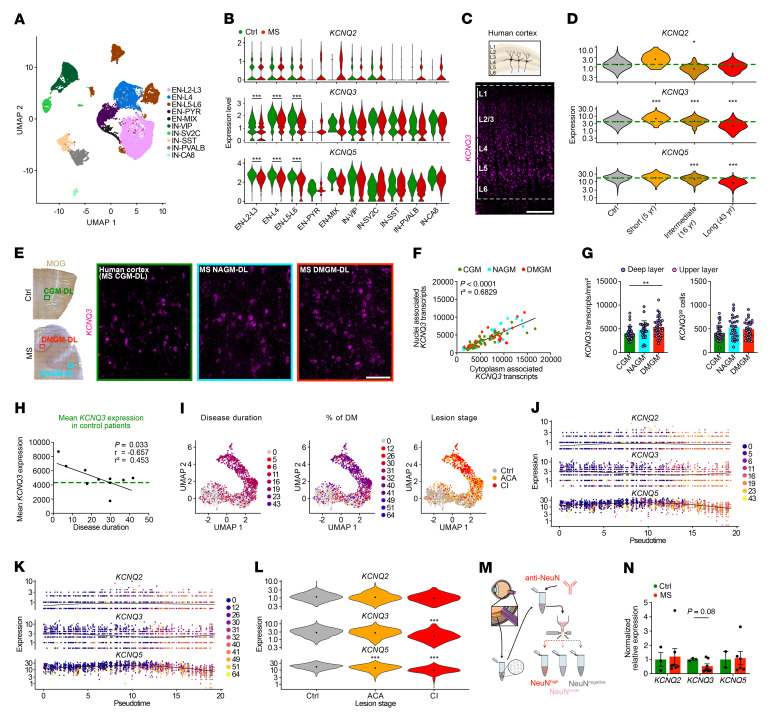
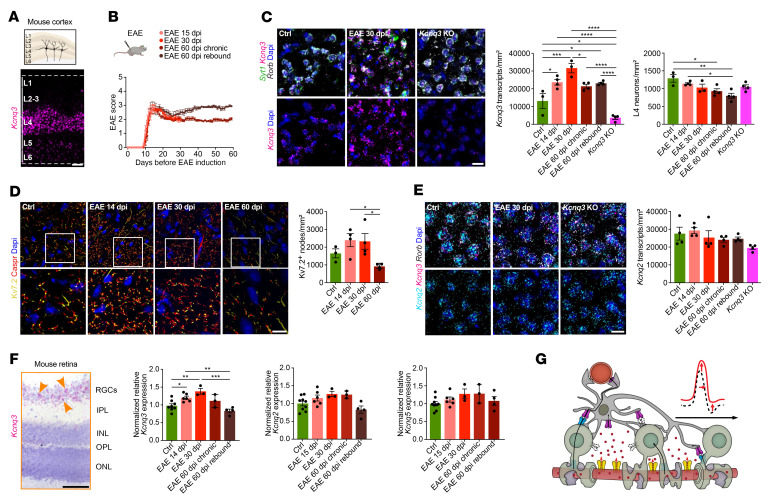
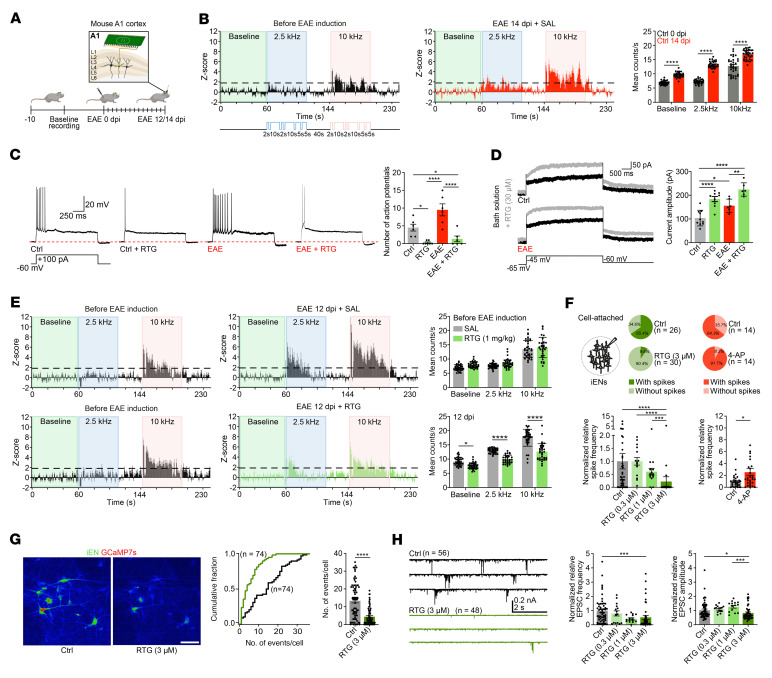
Comment in
-
Seeking neuroprotection in multiple sclerosis: an ongoing challenge.J Clin Invest. 2023 Apr 3;133(7):e168595. doi: 10.1172/JCI168595. J Clin Invest. 2023. PMID: 37009896 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
