

maxATAC: Genome-scale transcription-factor binding prediction from ATAC-seq with deep neural networks
- PMID: 36719906
- PMCID: PMC9917285
- DOI: 10.1371/journal.pcbi.1010863
maxATAC: Genome-scale transcription-factor binding prediction from ATAC-seq with deep neural networks
Abstract
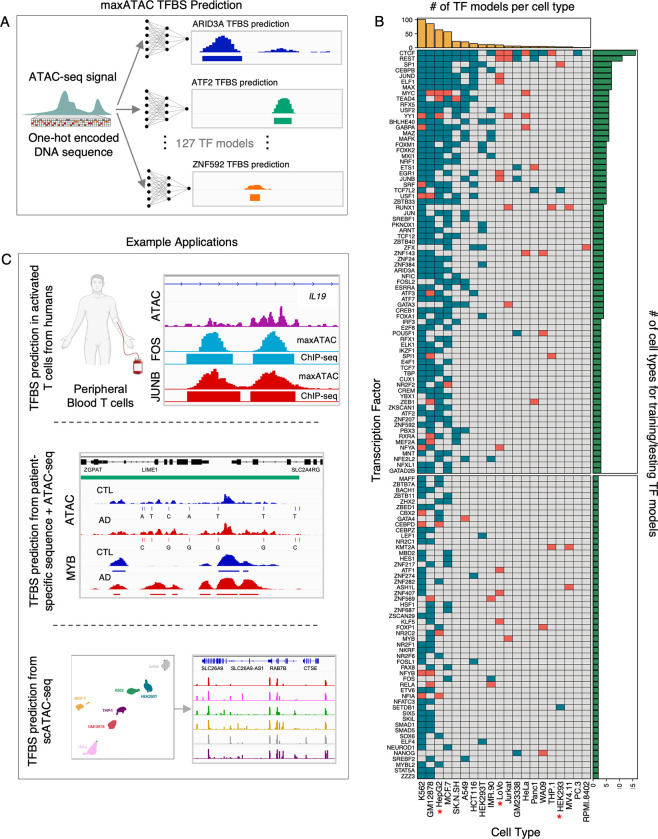
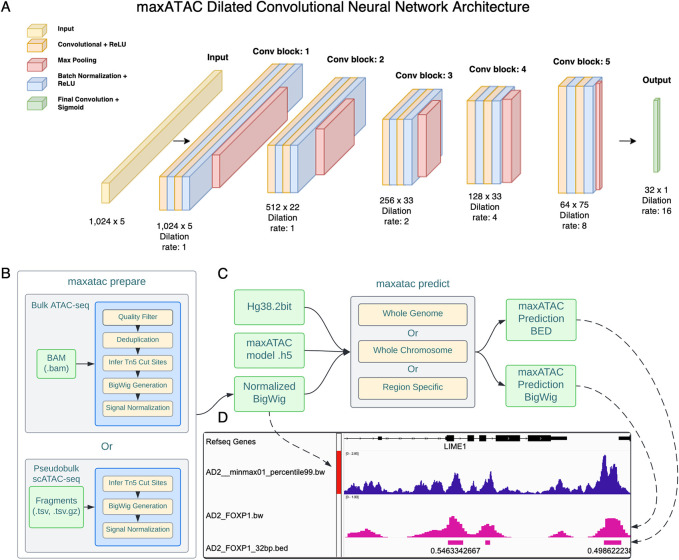
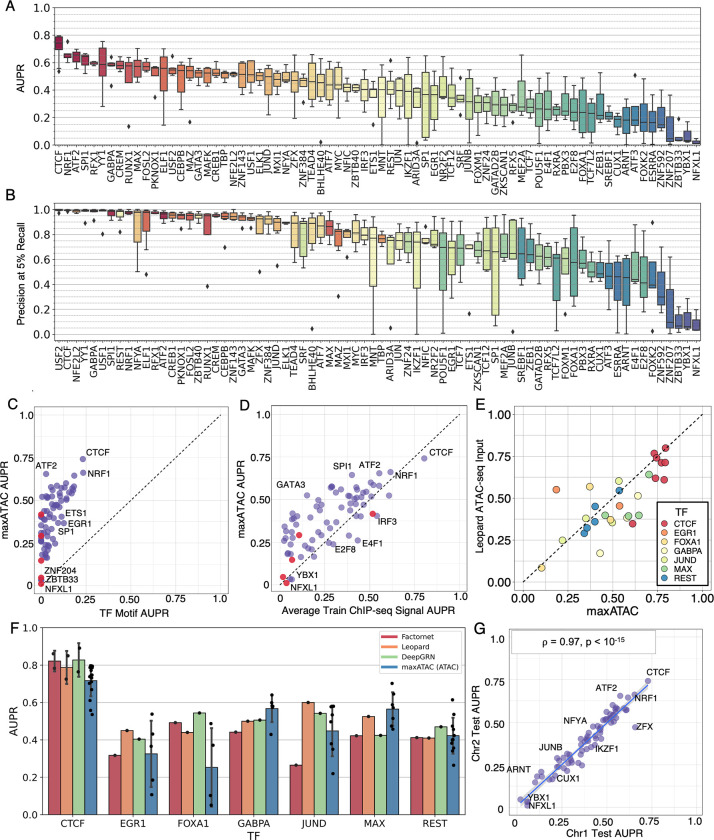
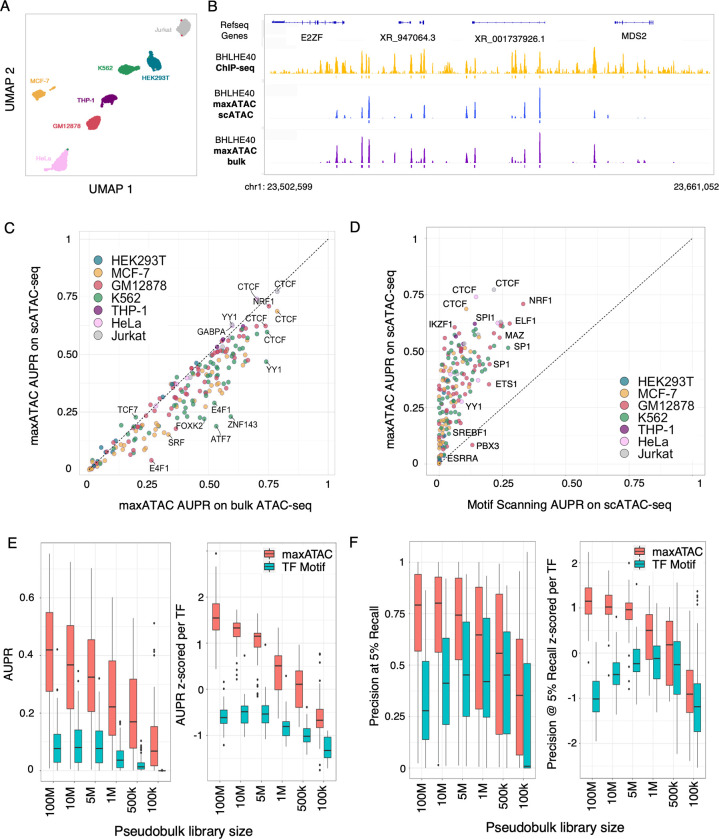
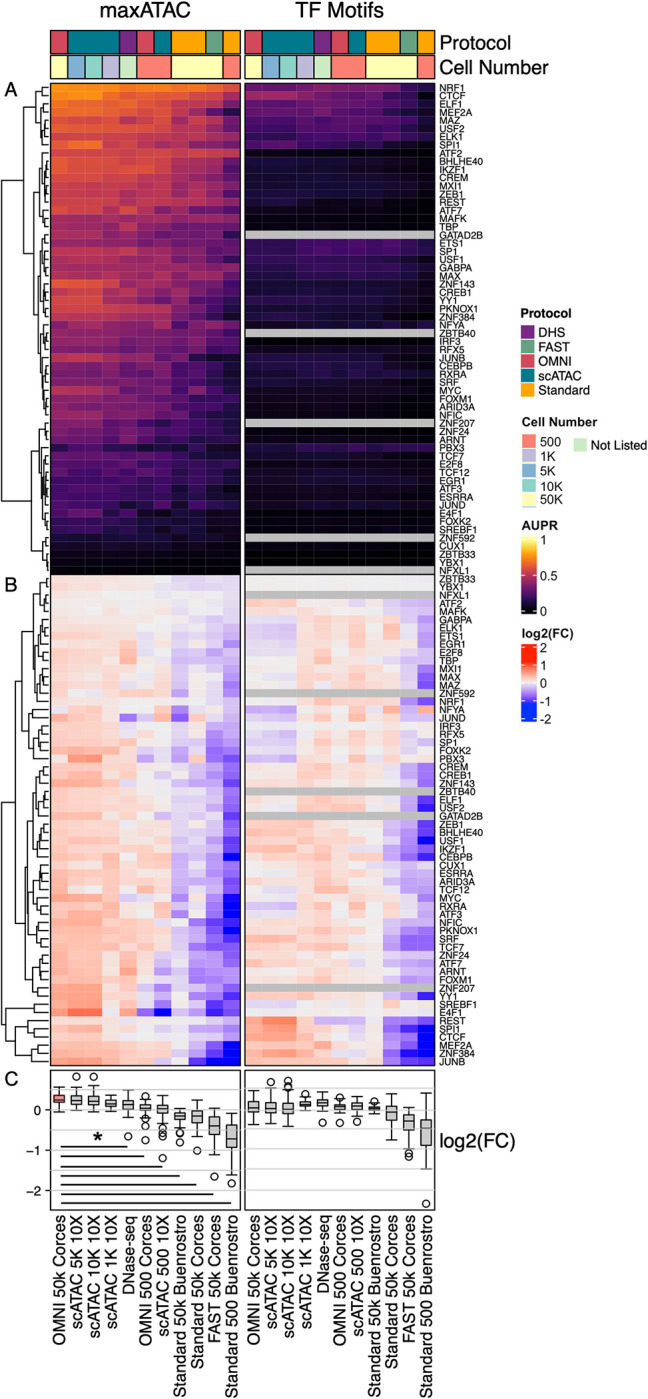
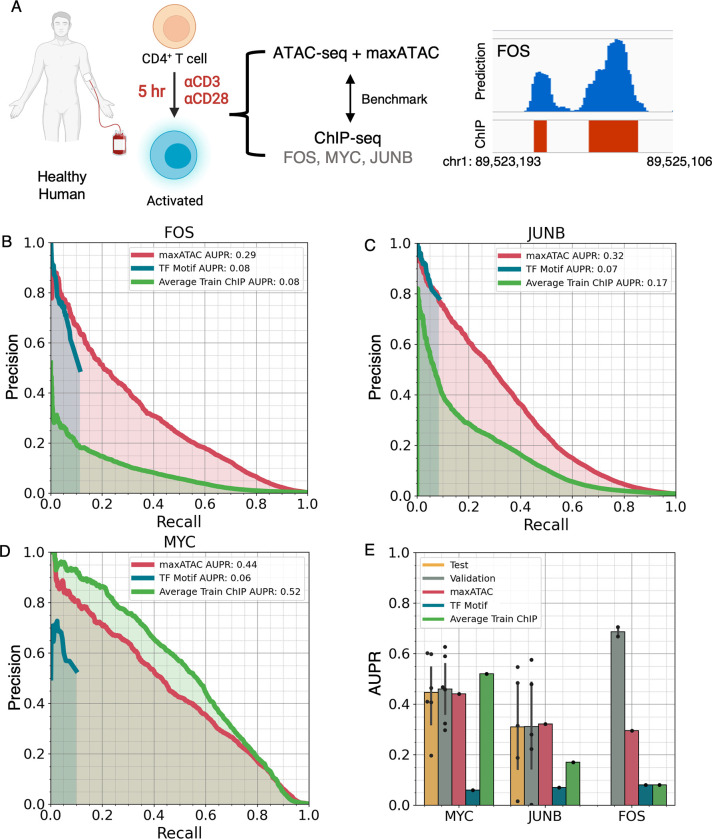
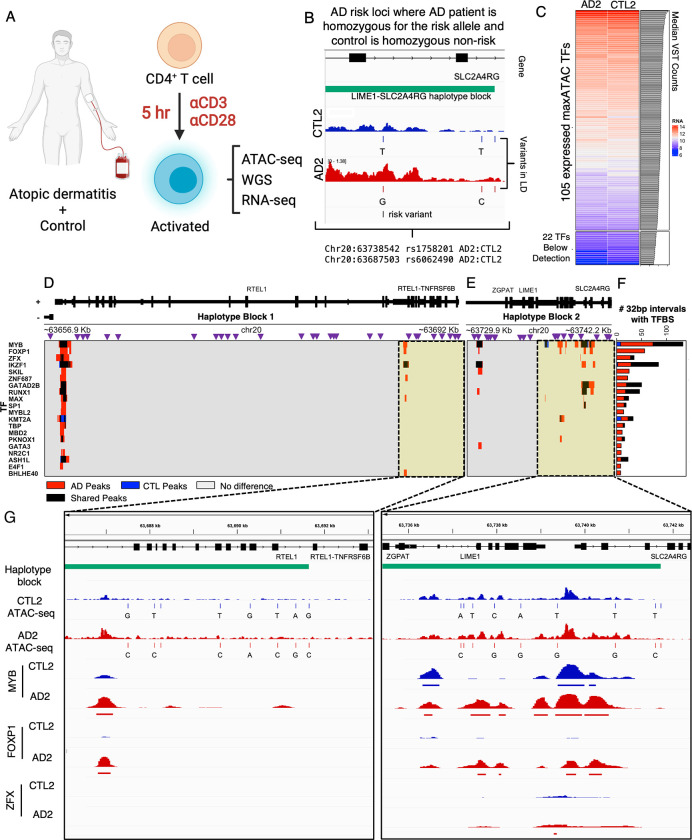
Transcription factors read the genome, fundamentally connecting DNA sequence to gene expression across diverse cell types. Determining how, where, and when TFs bind chromatin will advance our understanding of gene regulatory networks and cellular behavior. The 2017 ENCODE-DREAM in vivo Transcription-Factor Binding Site (TFBS) Prediction Challenge highlighted the value of chromatin accessibility data to TFBS prediction, establishing state-of-the-art methods for TFBS prediction from DNase-seq. However, the more recent Assay-for-Transposase-Accessible-Chromatin (ATAC)-seq has surpassed DNase-seq as the most widely-used chromatin accessibility profiling method. Furthermore, ATAC-seq is the only such technique available at single-cell resolution from standard commercial platforms. While ATAC-seq datasets grow exponentially, suboptimal motif scanning is unfortunately the most common method for TFBS prediction from ATAC-seq. To enable community access to state-of-the-art TFBS prediction from ATAC-seq, we (1) curated an extensive benchmark dataset (127 TFs) for ATAC-seq model training and (2) built "maxATAC", a suite of user-friendly, deep neural network models for genome-wide TFBS prediction from ATAC-seq in any cell type. With models available for 127 human TFs, maxATAC is the largest collection of high-performance TFBS prediction models for ATAC-seq. maxATAC performance extends to primary cells and single-cell ATAC-seq, enabling improved TFBS prediction in vivo. We demonstrate maxATAC's capabilities by identifying TFBS associated with allele-dependent chromatin accessibility at atopic dermatitis genetic risk loci.
Copyright: © 2023 Cazares et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: AB is a co-founder of Datirium, LLC.
Figures
References
-
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al.. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences. 2009;106: 9362–9367. doi: 10.1073/pnas.0903103106 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 AI130830/AI/NIAID NIH HHS/United States
- R01 NS099068/NS/NINDS NIH HHS/United States
- R01 AI153442/AI/NIAID NIH HHS/United States
- R01 DK107502/DK/NIDDK NIH HHS/United States
- U01 AI150748/AI/NIAID NIH HHS/United States
- R21 AI156185/AI/NIAID NIH HHS/United States
- R01 HG010730/HG/NHGRI NIH HHS/United States
- R01 GM055479/GM/NIGMS NIH HHS/United States
- R01 AI024717/AI/NIAID NIH HHS/United States
- P30 AR070549/AR/NIAMS NIH HHS/United States
- R01 AI148276/AI/NIAID NIH HHS/United States
- U19 AI070235/AI/NIAID NIH HHS/United States
- P01 AI150585/AI/NIAID NIH HHS/United States
- R01 AR073228/AR/NIAMS NIH HHS/United States
- U01 HG011172/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
