

Spatially resolved transcriptomic profiling of degraded and challenging fresh frozen samples
- PMID: 36720873
- PMCID: PMC9889806
- DOI: 10.1038/s41467-023-36071-5
Spatially resolved transcriptomic profiling of degraded and challenging fresh frozen samples
Abstract
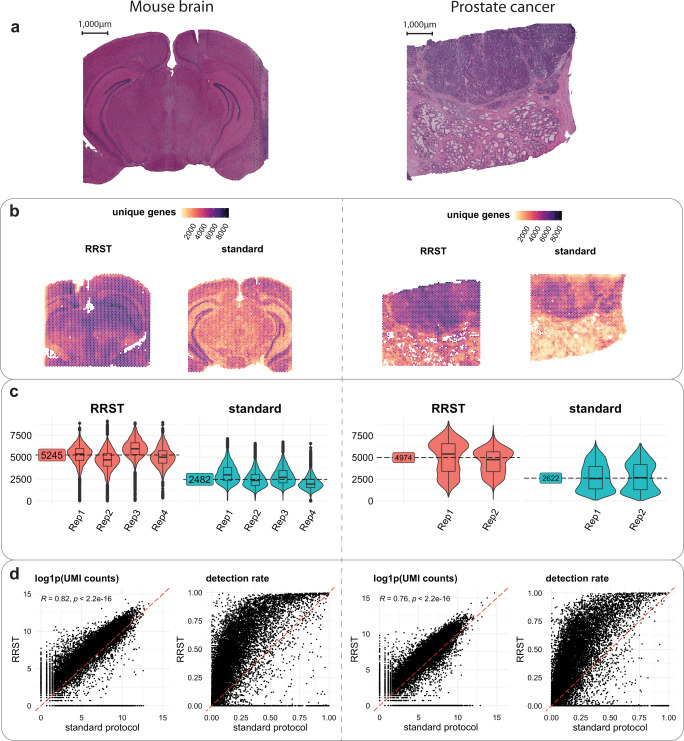
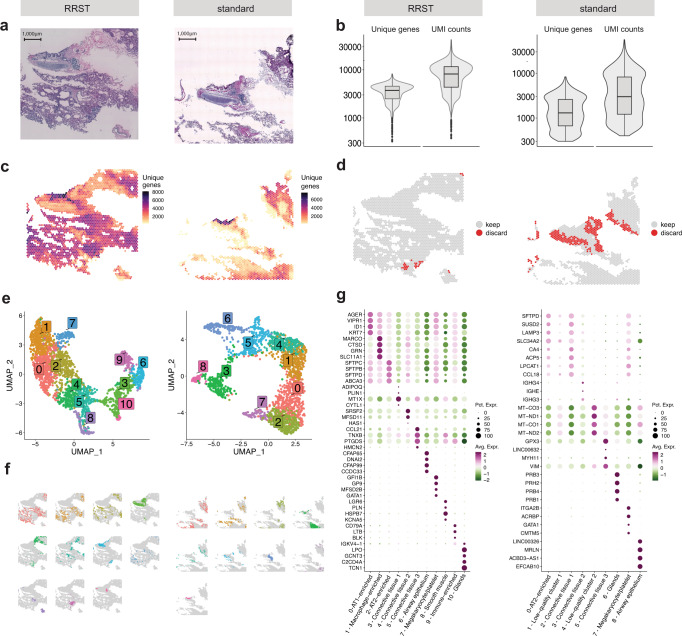
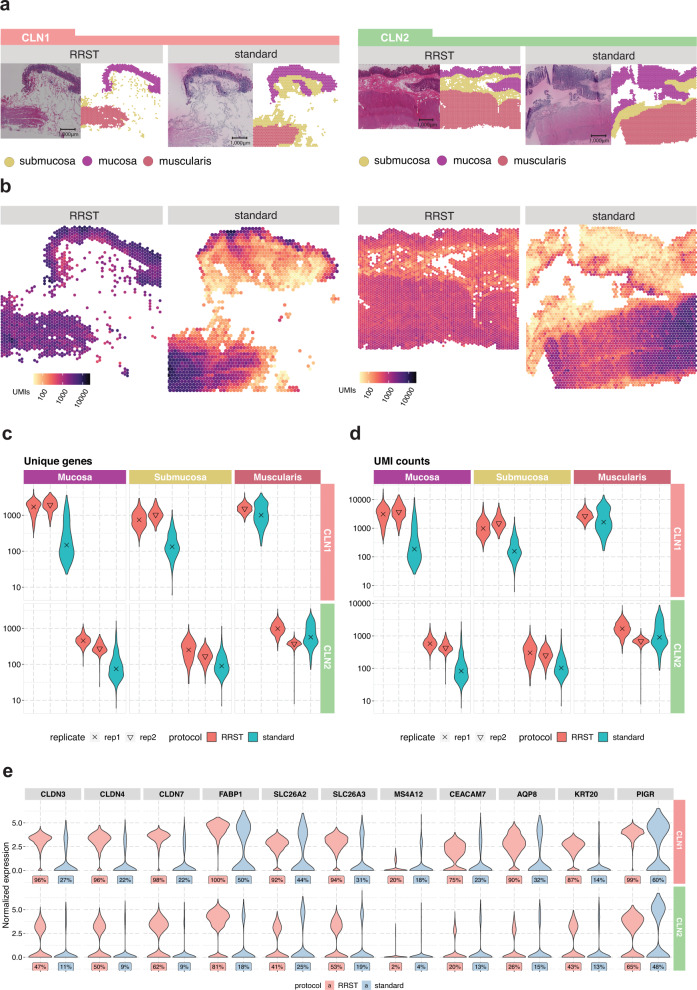
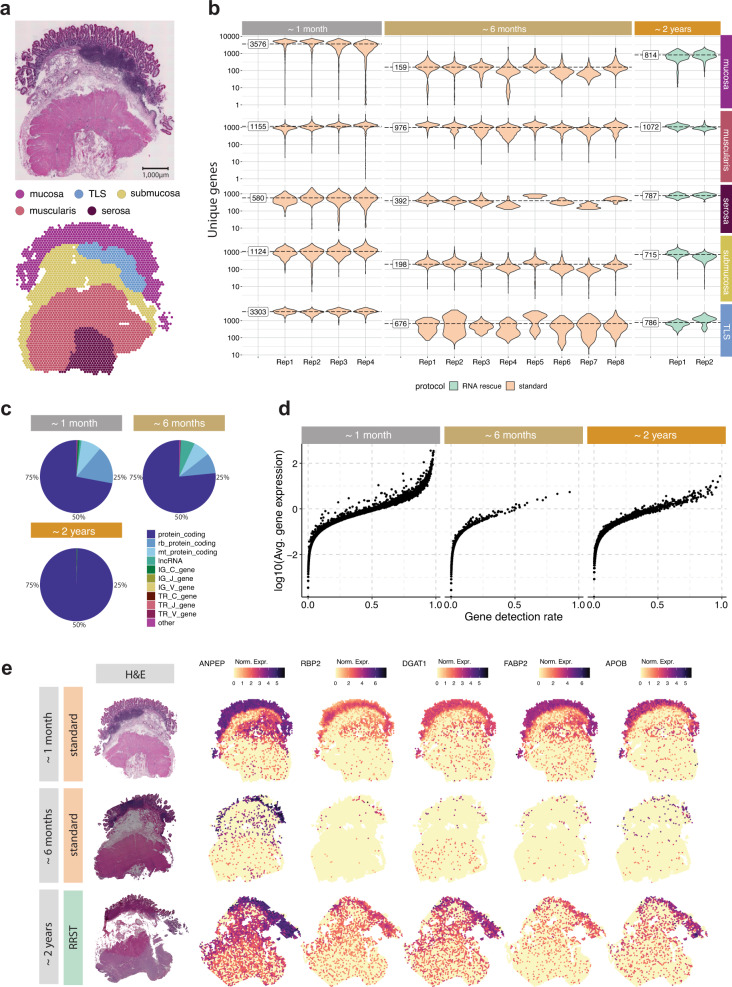
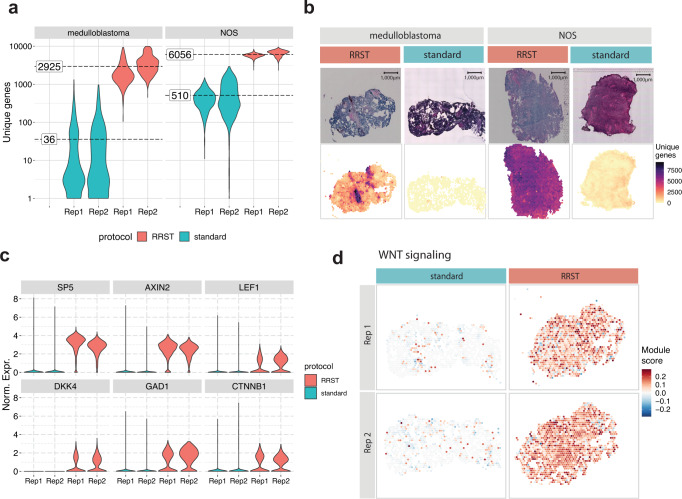
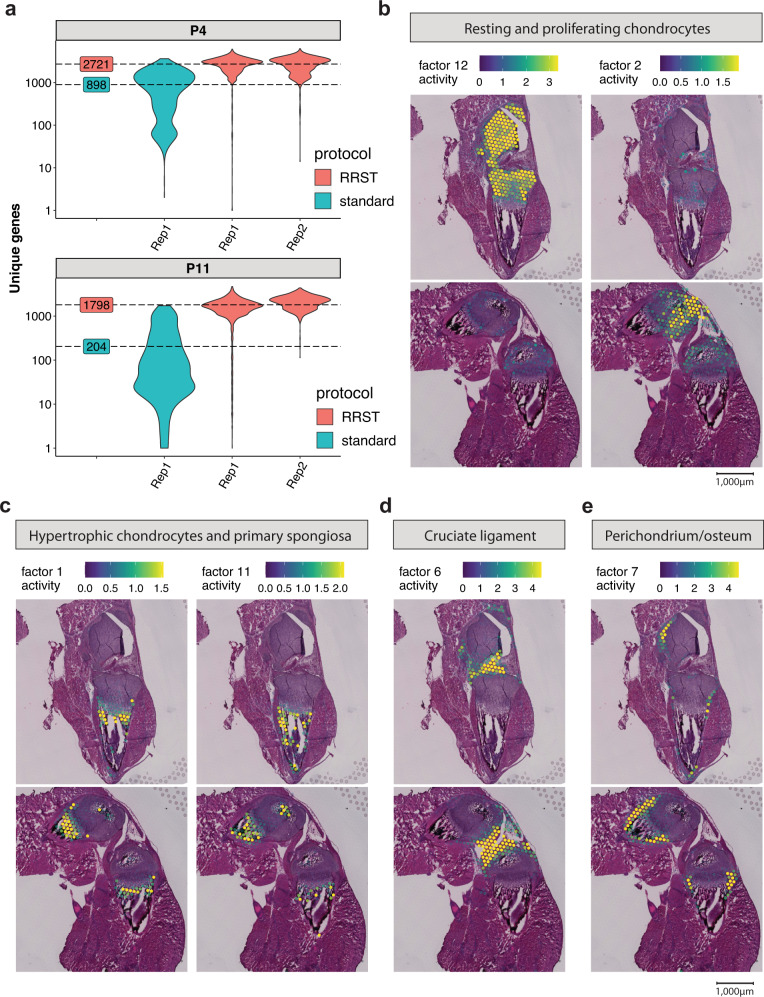
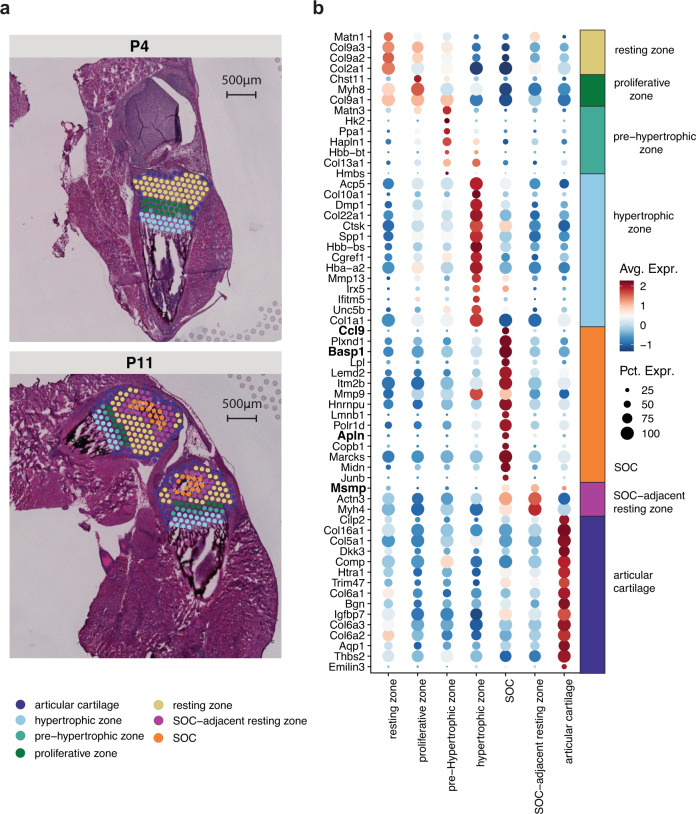
Spatially resolved transcriptomics has enabled precise genome-wide mRNA expression profiling within tissue sections. The performance of methods targeting the polyA tails of mRNA relies on the availability of specimens with high RNA quality. Moreover, the high cost of currently available spatial resolved transcriptomics assays requires a careful sample screening process to increase the chance of obtaining high-quality data. Indeed, the upfront analysis of RNA quality can show considerable variability due to sample handling, storage, and/or intrinsic factors. We present RNA-Rescue Spatial Transcriptomics (RRST), a workflow designed to improve mRNA recovery from fresh frozen specimens with moderate to low RNA quality. First, we provide a benchmark of RRST against the standard Visium spatial gene expression protocol on high RNA quality samples represented by mouse brain and prostate cancer samples. Then, we test the RRST protocol on tissue sections collected from five challenging tissue types, including human lung, colon, small intestine, pediatric brain tumor, and mouse bone/cartilage. In total, we analyze 52 tissue sections and demonstrate that RRST is a versatile, powerful, and reproducible protocol for fresh frozen specimens of different qualities and origins.
© 2023. The Author(s).
Conflict of interest statement
R.M., Z.A., L.L., L.A.G., X.A., L.K., and J.L. are scientific consultants for 10× Genomics, which holds IP rights to the ST technology. The remaining authors declare no competing interests.
Figures
References
-
- Larsson L, Frisén J, Lundeberg J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods. 2021;18:15–18. - PubMed
-
- Moses L, Pachter L. Museum of spatial transcriptomics. Nat. Methods. 2022;19:534–546. - PubMed
-
- Maniatis S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93. - PubMed
-
- Asp M, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179:1647–1660.e19. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
