

Molecular Evolution of Classic Hodgkin Lymphoma Revealed Through Whole-Genome Sequencing of Hodgkin and Reed Sternberg Cells
- PMID: 36723991
- PMCID: PMC10150291
- DOI: 10.1158/2643-3230.BCD-22-0128
Molecular Evolution of Classic Hodgkin Lymphoma Revealed Through Whole-Genome Sequencing of Hodgkin and Reed Sternberg Cells
Abstract
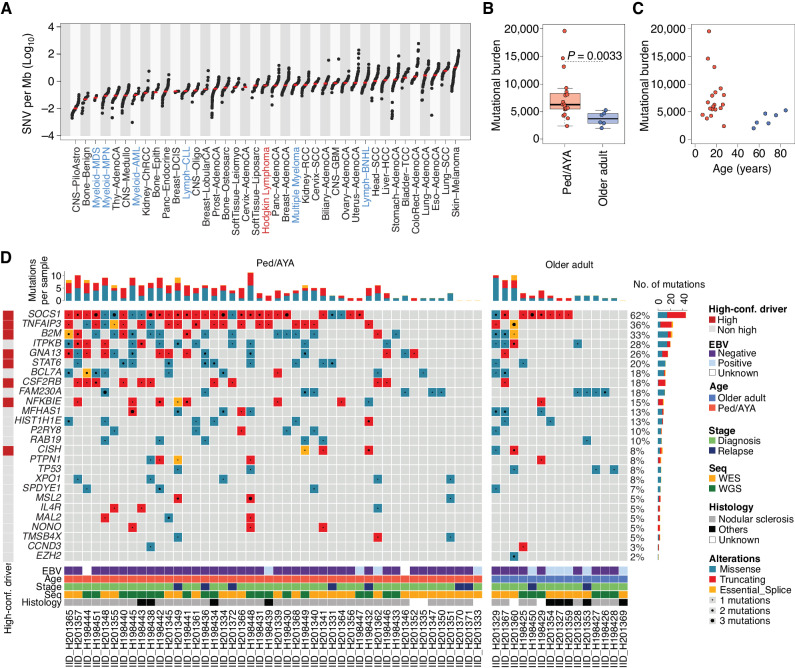
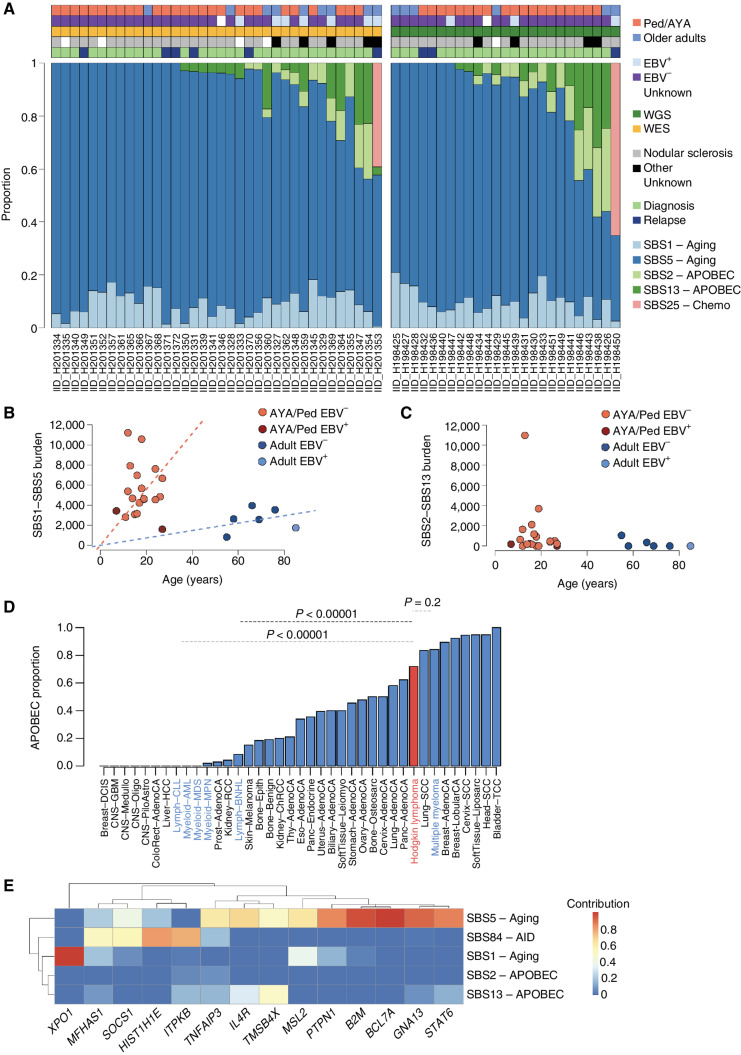
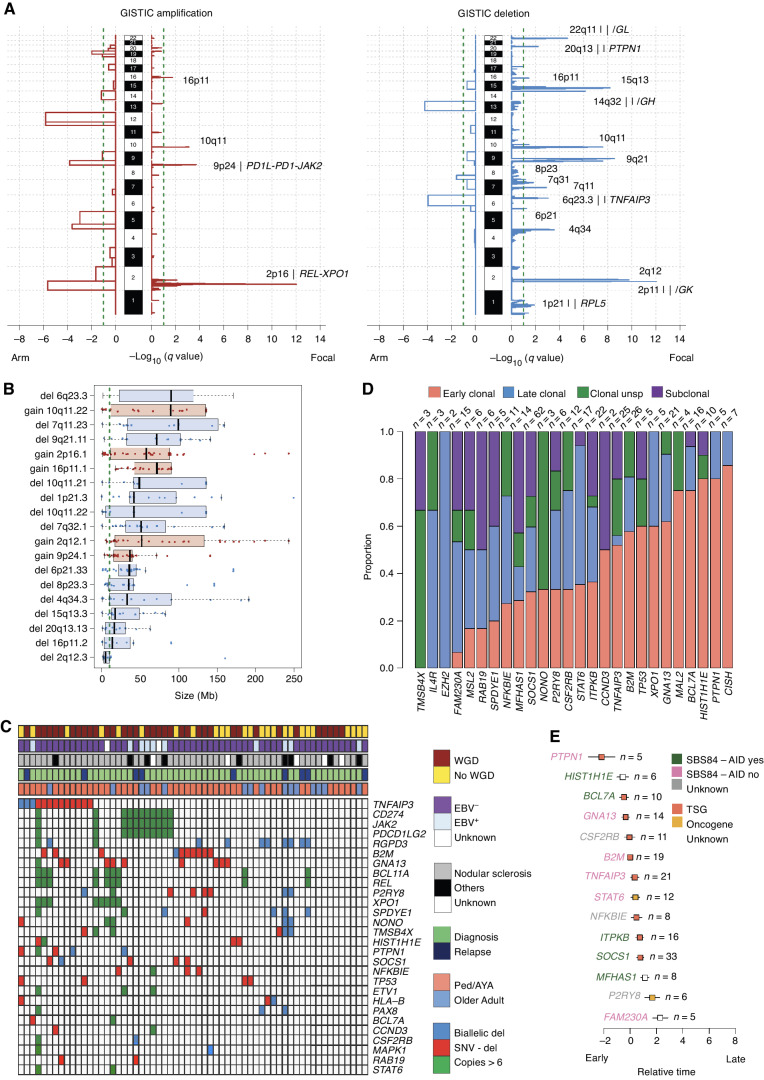
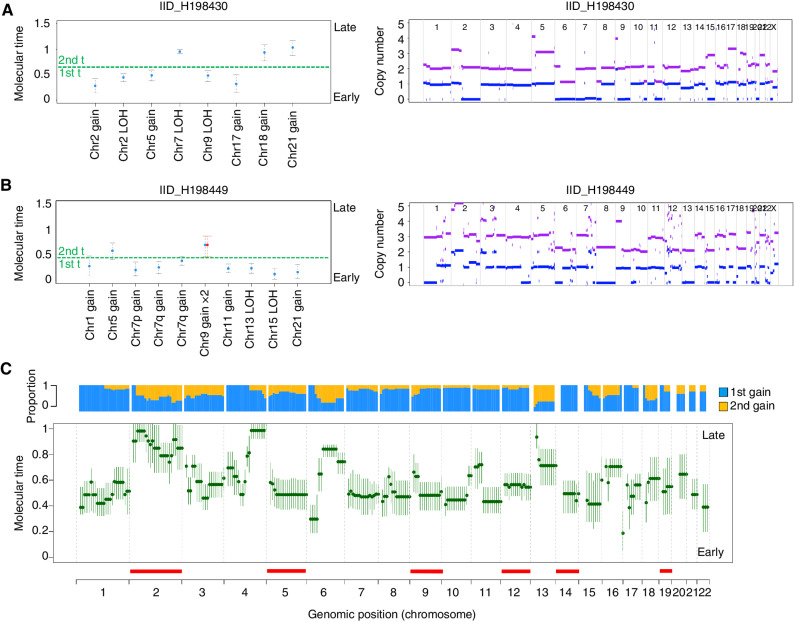
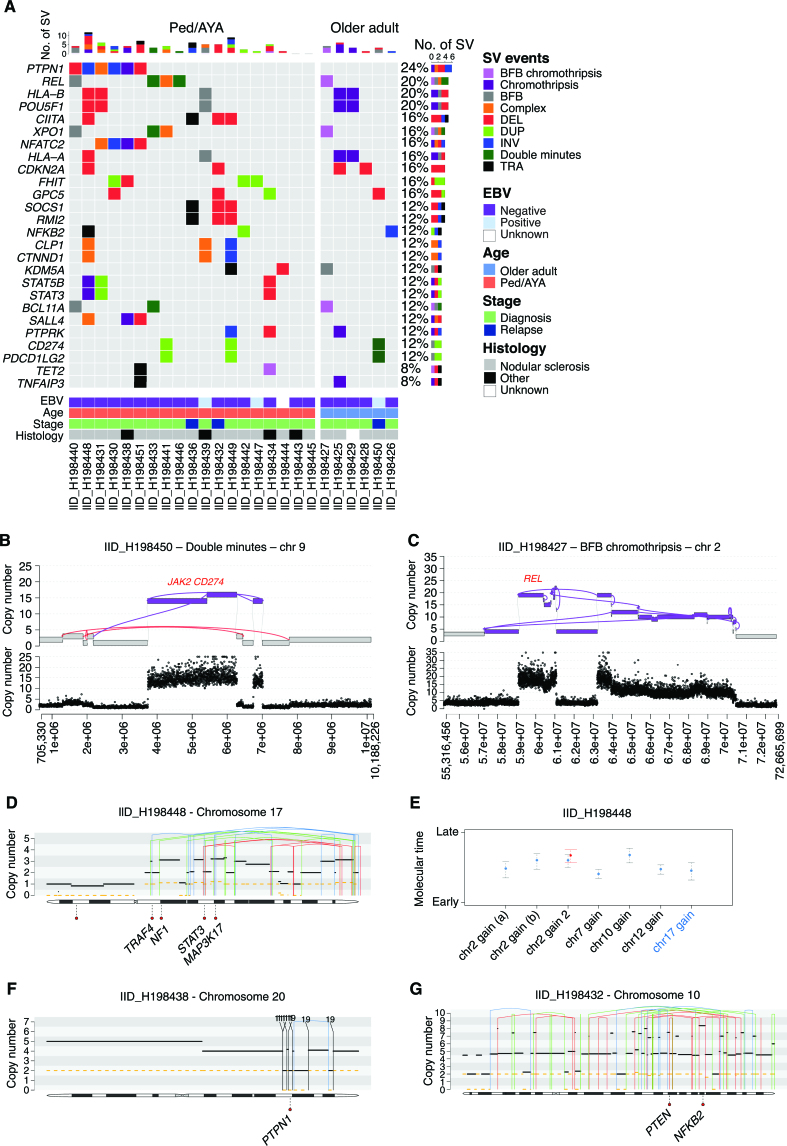
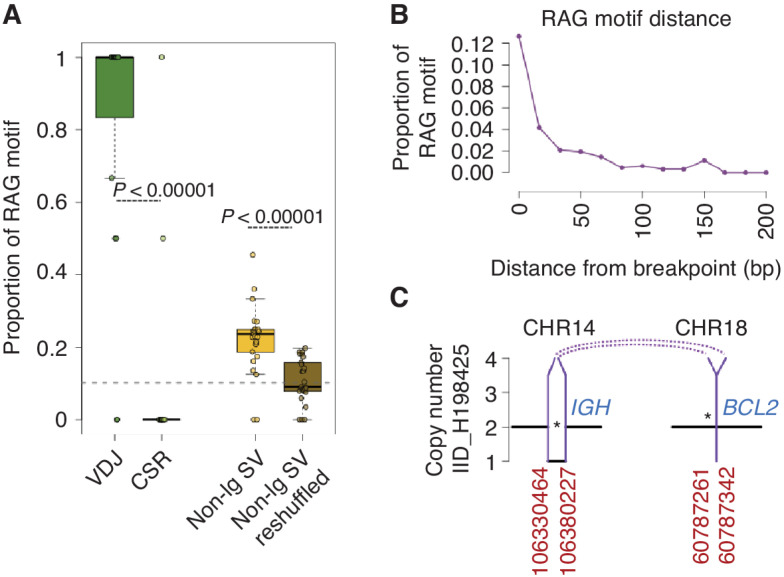
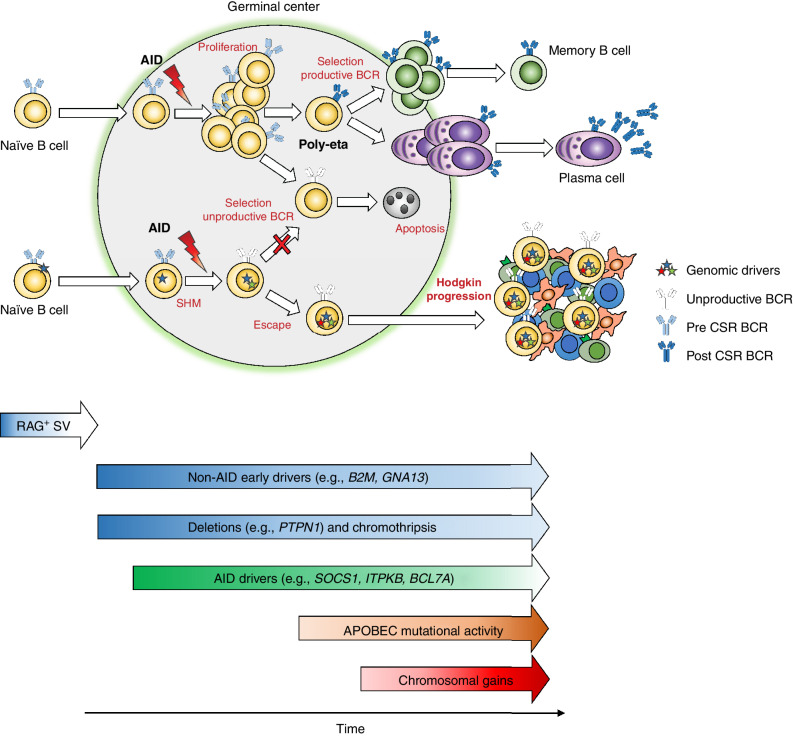
The rarity of malignant Hodgkin and Reed Sternberg (HRS) cells in classic Hodgkin lymphoma (cHL) limits the ability to study the genomics of cHL. To circumvent this, our group has previously optimized fluorescence-activated cell sorting to purify HRS cells. Using this approach, we now report the whole-genome sequencing landscape of HRS cells and reconstruct the chronology and likely etiology of pathogenic events leading to cHL. We identified alterations in driver genes not previously described in cHL, APOBEC mutational activity, and the presence of complex structural variants including chromothripsis. We found that high ploidy in cHL is often acquired through multiple, independent chromosomal gains events including whole-genome duplication. Evolutionary timing analyses revealed that structural variants enriched for RAG motifs, driver mutations in B2M, BCL7A, GNA13, and PTPN1, and the onset of AID-driven mutagenesis usually preceded large chromosomal gains. This study provides a temporal reconstruction of cHL pathogenesis.
Significance: Previous studies in cHL were limited to coding sequences and therefore not able to comprehensively decipher the tumor complexity. Here, leveraging cHL whole-genome characterization, we identify driver events and reconstruct the tumor evolution, finding that structural variants, driver mutations, and AID mutagenesis precede chromosomal gains. This article is highlighted in the In This Issue feature, p. 171.
©2023 American Association for Cancer Research.
Figures
Comment in
- 2643-3230. doi: 10.1158/2643-3230.BCD-4-3-ITI doi: 10.1158/2643-3230.BCD-4-3-ITI
References
-
- Mathas S, Hartmann S, Kuppers R. Hodgkin lymphoma: pathology and biology. Semin Hematol 2016;53:139–47. - PubMed
-
- Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015;125:1061–72. - PubMed
-
- Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010;116:3268–77. - PMC - PubMed
-
- Joos S, Kupper M, Ohl S, von Bonin F, Mechtersheimer G, Bentz M, et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res 2000;60:549–52. - PubMed
-
- Joos S, Menz CK, Wrobel G, Siebert R, Gesk S, Ohl S, et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002;99:1381–7. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
